Obstacles and innovations of macrocyclic drug development
Posted: 16 January 2025 | Ann E Cleves (VP of Application Science for Optibrium’s BioPharmics Division) | No comments yet
Macrocycles outperform traditional therapeutics on several fronts, yet they account for just four percent of FDA-approved drugs. Colleagues Ann E Cleves and Ajay N Jain from Optibrium Ltd explain why we can expect to see this number increase in the coming years.

What are macrocycles and why are they interesting for drug discovery?
Traditionally, drug discovery has focused on small-molecule therapeutics, typically with a molecular weight of less than 500 Daltons.1 However, there has recently been an increased interest in so-called ‘beyond rule-of-five’ compounds, such as macrocycles. While small molecules typically contain rings of five to seven atoms, which are relatively inflexible, macrocycles usually contain 12 or more atoms in a ring, although they can be significantly larger (Figure 1). The larger rings within these molecules create a higher degree of flexibility, enabling them to adopt a large range of conformations. Their larger surface area also allows greater interaction with the target, thus they bind to their targets with higher affinity and selectivity than many non-macrocyclic drugs.2
Typically, small-molecule drugs target active sites buried inside proteins. However, some active sites are large, flat sites or shallow grooves on the surface of the protein. These sites are commonly involved in protein-protein interactions, some of which are key to disease development. Many of these proteins were considered undruggable, as small molecules bind to such sites with very low affinity.3
Antibodies are primarily used to target extracellular proteins, though researchers have generated antibodies that can reach intracellular targets. However, it remains challenging to design antibodies that cross cell membranes. Macrocycles, by contrast, have a higher degree of cell permeability and are also capable of binding to protein surface sites due to their size and flexibility.4 Macrocyclic compounds have also been shown to disrupt protein-protein interactions, highlighting their potential as therapeutic agents.3
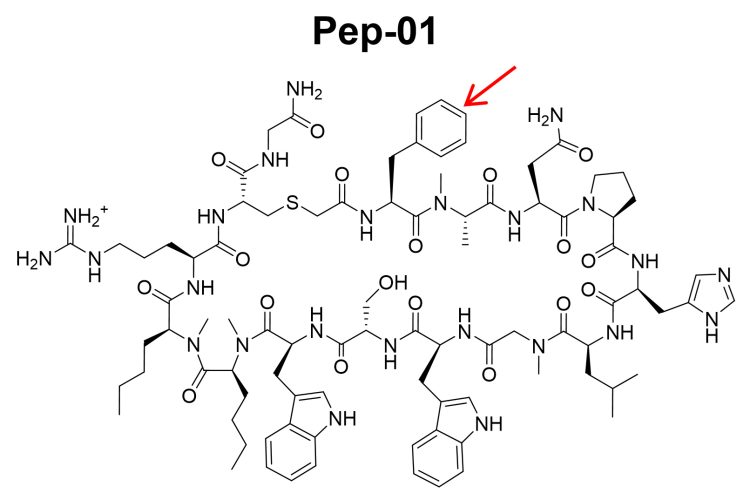
Figure 1: Structure of Pep-01, a large macrocycle investigated as a cancer therapeutic targeting the PD1-PDL1 interface.5
Macrocycles as drugs
Despite accounting for just four percent of the U.S. Food and Drug Administration (FDA) approved drugs, macrocycles are used to combat a range of diseases, including cancer, viral, fungal and bacterial infections, neurodegenerative and autoimmune diseases.6 Among the well-known FDA-approved macrocyclic drugs are those that treat Hepatitis C, a disease with previously limited therapeutic options. These drugs, including grazoprevir and vaniprevir, were the result of macrocyclisation of acyclic precursors.7 Another more recently developed macrocyclic drug is the oral PCSK9 inhibitor MK-0616, which Merck produced to combat atherosclerotic cardiovascular disease. Phase III trials for MK-0616 commenced in the second half of 2023.8
Why are there not more macrocyclic drugs?
The focus on small molecules in drug discovery was driven in part by the ‘Rule of Five’ (Ro5), a set of guidelines designed to help identify compounds with an increased likelihood of high oral absorption.1 However, the Ro5 was derived from observations of the characteristics of previously discovered small-molecule drugs, and an overreliance on these guidelines may have led to opportunities being overlooked in lesser explored areas of chemistry. 9,10
Because many therapeutic macrocycles violate the Ro5 guidelines, the presumption was that such large molecules would have limited cell permeability. However, close to 40 percent of all macrocyclic drugs are orally bioavailable,6 including natural products and derivatives thereof that have provided chemical structure clues with respect to permeability. Researchers have shown that cell permeability can be improved by designing macrocyclic peptides whose structure includes amide (NH) groups that are engaged in internal hydrogen bonding interactions.11 Macrocycles can also be engineered in other ways to become more cell permeable; for example, by incorporating N-methyl groups in macrocyclic peptides.12 Ring bridging strategies have also been effective in addressing permeability, as seen with MK-0616.8 Furthermore, oral bioavailability is not always essential; for example, in disease areas such as cancer, intravenous administration of drugs is common.
Metabolism was another concern in the early development of macrocyclic peptides. Many peptides contain natural amino acids that are easily metabolised, resulting in rapid clearance thereby limiting their application as therapeutics. However, research has shown that constrained cyclic peptides, like macrocycles, are often more stable than non-cyclic peptides.13 It is also possible to improve metabolic properties by modifying the macrocyclic peptide backbone with incorporation of non-natural amino acids and N-methylation.14
How the discovery process differs for macrocycles
The initial process of discovery for small molecules and large macrocycles is similar, but later stages in discovery, such as optimisation and synthesis, are trickier due to difficulties working with larger and more flexible molecules. Key challenges include synthetic modification, assay execution, scalable synthesis, crystal structure solution, and especially computational modelling. Predicting how a molecule binds to a protein, what it looks like in solution and modelling its energetics is significantly more difficult with large flexible molecules. While computer-aided drug design (CADD) has enabled accurate modelling of many aspects of small molecules for many years, the greater size and flexibility of macrocycles has posed a major barrier to applying CADD to this class of molecules.
However, the recent development of more efficient and generalisable 3D conformation generation methods has changed this, enabling the vast numbers of potential conformations that macrocycles can adopt to be explored via CADD.15 In addition, cross-disciplinary approaches that marry computational approaches with experimental ones can help ameliorate the conformational challenges. Figure 2 shows how computational conformational exploration can be done in the context of experimental nuclear magnetic resonance (NMR) data to produce a tractable macrocyclic solution ensemble that mirrors the way in which the molecule binds its target protein.5
Another important difference in discovery is the impact of molecular strain on the design process. Strain is the energy needed to force a molecule into a particular 3D shape or conformation. For macrocycles, the potential for large surface contact area with the target allows them to bear more strain than small molecules.16 But a complicating factor is that conformational strain for these molecules can differ greatly when even small changes are made to the molecular structure. In Pep-01, if the phenylalanine (red arrow, Figure 1) is replaced by alanine, a difference of just a few atoms, the binding affinity is dramatically reduced. This effect is due to a change in molecular strain rather than a change in the interaction with the protein.5 New computational models of strain present opportunities for designing lower-strain bound conformations16 and, combined with conventional CADD methods for assessing binding interactions with a protein, guide the optimisation of high-affinity macrocycles.
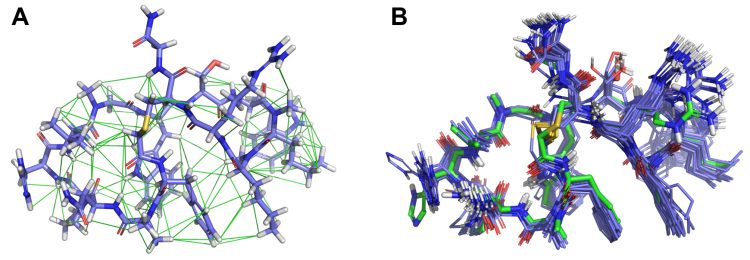
Figure 2: (A) Pep-01 (blue) in its lowest-energy solution conformation that satisfies experimental NMR inter-proton restraints (green); and (B) a comparison of the full NMR-restrained solution ensemble (blue) to the PD-L1-bound form (green). In consideration of experimental NMR restraints, biologically relevant conformations can be identified, reflecting macrocyclic behaviour in solution which often matches what is observed when binding the biological target.
Streamlining macrocyclic therapeutic development
Platforms such as PeptiDream’s drug discovery platform system17 have facilitated easier synthesis and screening of macrocyclic peptide libraries for lead compounds (eg, the PD-L1 macrocycle from Figure 1). Another approach to macrocyclic library design is to generate pools of peptidic scaffolds that are designed to possess qualities such as permeability, potentially obviating the need for major scaffold changes.18
Given the challenges around macrocycle flexibility, obtaining structural information on both macrocycles and target proteins is essential to understanding binding conformations and affinities. As discussed, the degree of flexibility of large macrocycles means that binding conformations must be identified from a very large range of possible conformations. As shown earlier, computational models can use data from biophysical experiments such as NMR. Improvements in modelling the bound state of macrocycles in modalities such as X-ray crystallography and cryogenic electron microscopy (cryo-EM) can significantly aid identifying low-strain conformational ensembles that correspond to the bound form.16
Modern biophysical tools and crystallographic techniques can provide us with a wealth of data, but making the best use of the data may require paradigm shifts. Figure 3 shows the contrast between the traditional method of fitting a macrocyclic ligand into X-ray density (left), which makes use of a single conformation, and a newer approach (right) that fits an ensemble of conformations.19,20 The traditional fitting approach has a limited model of strain and it models atomic coordinate uncertainty using a temperature-based concept, with atoms matching poor electron density as being “hot” (red in Figure 3). Newer methods model uncertainty with a collection of low-strain conformers (orange in Figure 3), leading to better fit while also dramatically reducing the estimated strain of the bound state of the macrocycle.19,16 Advances in CADD have complemented the increasing availability of quality structural data, facilitating transformative insights into the structure of macrocyclic compounds and how these interact with target proteins.
The marriage of cross-disciplinary teams of biophysical and computational scientists enables discovery and optimisation of better macrocycle molecules, supporting more efficient macrocyclic drug discovery.5
n
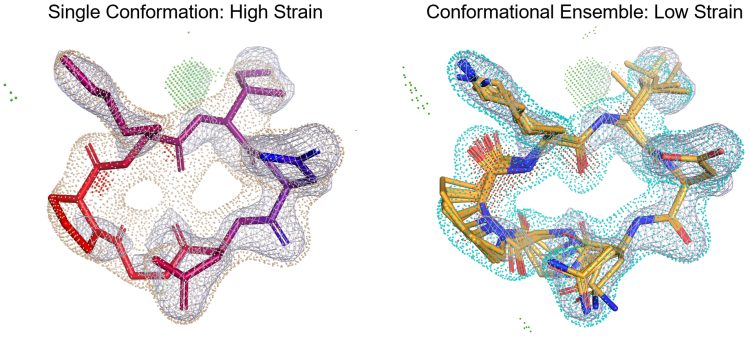
Figure 3: A demonstration of the improvements in modelling the fit of macrocycle ligand conformations into X-ray density on a known protein-ligand complex (3WNE PDB). By considering ensembles instead of single conformers, it is possible to reduce the estimated strain of the complex, and to more realistically model how a ligand interacts with a target protein.
Conclusion
Due in part to an over-adherence to the Ro5, macrocycles have not previously been a go-to compound class for drug design. However, these molecules are becoming increasingly popular in drug discovery due to their ability to target previously undruggable proteins. They present a distinct solution to targeting those proteins that are not tractable using other groups of drugs, such as small molecules or antibodies. Recent developments in peptide screening platforms, advancements in CADD, and the ability to apply information gained from biophysical experiments to molecular models have played a key role in making the development of macrocyclic drugs easier. Additionally, this field is supported by industry trends towards a more multi- and interdisciplinary approach. As computational chemists, medicinal chemists, biophysicists and biologists collaborate closely and combine biophysical data with computational methods, macrocyclic therapeutic development is becoming increasingly feasible and efficient.
About the authors

Ann E Cleves
Ann is VP of Application Science for Optibrium’s BioPharmics Division, which focuses on 3D computational modelling for molecular design. She has a PhD in Microbiology and Cell Biology from the University of Illinois, followed by work in biochemistry as a post-doctoral fellow at UCSF.
 Ajay N Jain
Ajay N Jain
Ajay is VP of Research for the Optibrium’s BioPharmics Division. He has a PhD in Computer Science from Carnegie Mellon University.
References
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced Drug Delivery Reviews, 64:4–17, 2012.
- Driggers EM, Hale SP, Lee J, Terrett NK. The exploration of macrocycles for drug discovery: An underexploited structural class. Nature Reviews Drug Discovery, 7(7):608–624, 2008.
- Dougherty PG, Qian Z, Pei D. Macrocycles as protein-protein interaction inhibitors. Biochemical Journal, 474(7):1109–1125, 2017.
- Heinis C. Tools and rules for macrocycles. Nature Chemical Biology, 10(9):696–698, 2014.
- Jain AN, Brueckner AC, Jorge C, et al. Complex peptide macrocycle optimization: Combining NMR restraints with conformational analysis to guide structure-based and ligand-based design. Journal of Computer-Aided Molecular Design, 37(11): 519–535, 2023.
- Jimenez DG, Poongavanam V, Kihlberg J. Macrocycles in drug discovery: Learning from the past for the future. Journal of Medicinal Chemistry, 66(8):5377–5396, 2023.
- McCauley JA, Rudd MT. The invention of grazoprevir: An HCV NS3/4a protease inhibitor. HCV: The Journey from Discovery to a Cure: Volume I, pages 355–387, 2019.
- Ballantyne CM, Banka P, Mendez G, et al. Phase 2b randomized trial of the oral PCSK9 inhibitor MK-0616. Journal of the American College of Cardiology, 81(16):1553–1564, 2023.
- Doak BC, Zheng J, Dobritzsch D, Kihlberg J. How beyond rule of 5 drugs and clinical candidates bind to their targets. Journal of Medicinal Chemistry, 59(6):2312–2327, 2016.
- Yusof I, Segall MD. Considering the impact drug-like properties have on the chance of success. Drug Discovery Today, 18(13-14):659–666, 2013.
- Bhardwaj G, O’Connor J, Rettie S, et al. Accurate de novo design of membrane-traversing macrocycles. Cell, 185(19):3520–3532, 2022.
- Hoang HN, Hill TA, Fairlie DP. Connecting hydrophobic surfaces in cyclic peptides increases membrane permeability. Angewandte Chemie, 133(15):8466–8471, 2021.
- Costa L, Sousa E, Fernandes C. Cyclic peptides in pipeline: What future for these great molecules? Pharmaceuticals, 16(7):996, 2023.
- Khatri B, Nuthakki VR, Chatterjee J. Cyclic Peptide Design, Chapter 2: Strategies to Enhance Metabolic Stabilities. Springer, 2019.
- Jain AN, Cleves AE, Gao Q, et al. Complex macrocycle exploration: Parallel, heuristic, and constraint-based conformer generation using ForceGen. Journal of Computer-Aided Molecular Design, 33:531–558, 2019.
- Jain AN, Brueckner AC, Cleves AE, et al. A distributional model of bound ligand conformational strain: From small molecules up to large peptidic macrocycles. Journal of Medicinal Chemistry, 66(3):1955–1971, 2023.
- Goto Y, Suga H. The RaPID platform for the discovery of pseudo-natural macrocyclic peptides. Accounts of Chemical Research, 54(18):3604–3617, 2021.
- Tanada M, Tamiya M, Matsuo A, et al. Development of orally bioavailable peptides targeting an intracellular protein: From a hit to a clinical KRAS inhibitor. Journal of the American Chemical Society, 145 (30):16610–16620, 2023.
- Jain AN, Cleves AE, Brueckner AC, et al. XGen: Real-space fitting of complex ligand conformational ensembles to X-ray electron density maps. Journal of Medicinal Chemistry, 63(18):10509–10528, 2020.
- Brueckner AC, Deng Q, Cleves AE, et al. Conformational strain of macrocyclic peptides in ligand–receptor complexes based on advanced refinement of bound-state conformers. Journal of Medicinal Chemistry, 64(6):3282–3298, 2021.
Related topics
Amino Acids, Analysis, Assays, Cancer research, Computational techniques, Crystallography, Drug Discovery, Drug Discovery Processes, Nuclear Magnetic Resonance (NMR)
Related conditions
Atherosclerotic Cardiovascular Disease, autoimmune diseases, Cancer, Hepatitis C, Neurodegenerative diseases
Related organisations
Optibrium, PeptiDream
Related people
Ajay N. Jain, Ann E. Cleves