

Translational proteomics: from the bench to the bedside
Posted: 13 December 2016 | Manuel Fuentes (Cancer Research Centre), Paula Díez (Cancer Research Centre) | 1 comment
Understanding how a cell responds to different stimuli producing diverse protein profiles can provide information about cell signalling pathways, generating more specific treatments…

The term was coined in 1997 by Marc Wilkins as a portmanteau of protein and genome1. One of the major characteristics of proteomes is their variability, depending on time and biological conditions, which provides a great dynamism to cellular states2,3. Therefore, not all genes (genome) present in a cell may be expressed and translated into functional proteins. Thus, profiling the specific proteins produced by a cell at a certain point in time (e.g. activated cell, pathological cell or apoptotic cell) may have a notorious impact in the clinical field. In addition, information about post-translational modifications’ (PTMs) status and subcellular localisation could help to achieve a complete knowledge of these differential protein profiles.
A wide breadth of knowledge about how a cell responds to different stimuli producing diverse protein profiles can provide detailed information about cell signalling pathways which can, in turn, help to generate more specific treatments, reducing disease effects.
The main challenge of studying the proteome is related to its intrinsic complexity. When compared with the genome, the proteins of a singular entity present more factors to be analysed since one gene can encode more than one protein and these can present post-translational modifications, a wide range of concentrations and interaction networks. In addition, several proteins are difficult to isolate. Nevertheless, proteomics is a powerful discipline to supplement genomics, producing a deeper understanding of cell biology and physiology.
Proteomics not only offers quantitative and qualitative information about protein expression, it also provides an insightful view into when and where proteins are produced and how they interact with other biomolecules or proteins. In relation to protein localisation, subcellular proteomics is a powerful strategy for reducing the complexity of the proteome and enabling novel proteins to be discovered.
The Human Proteome Project (HPP), an international initiative, was organised by the Human Proteome Organization (HUPO) and aims to generate a comprehensive map of all proteins within the human body. Its principal purpose is to elucidate molecular and biological functions and so enable advances to be made in the diagnosis and treatment of diseases. The HPP has been divided into two general projects: chromosome-based HPP (C-HPP) and biology/disease HPP (B/D-HPP)4.
C-HPP is pursuing the systematic characterisation of proteins codified by each of the 23 pairs of chromosomes present in human cells, especially those proteins with PTMs, sequence variants and splice isoforms. To this end, the C-HPP integrates the information obtained by cell biology, biochemistry, metabolomics, molecular biology, bioinformatics, transcriptomics, genomics, physiology, clinical data and epigenomics5–7. Meanwhile, the B/D-HPP initiative provides profiles of relevant proteins in diseases or biological systems that have been obtained from coordinated studies from biology and/or disease8,9.
Proteomics technologies In general, proteome characterisation is performed by mass-spectrometry (MS) and affinity reagents-based techniques (such as antibody-based techniques to perform immunoassays) (Figure 1). The immunoassays have been traditionally used for the detection of proteins through enzyme-linked immunosorbent assays (ELISAs) or western blots (WB), among others. However, these approaches do not involve assays in a high-throughput format and small-medium density.
Novel approaches such as protein microarrays that incorporate (among other biomolecules) the usage of antibodies for the profiling of proteomes are flourishing. The major advantage of these microarrays is their versatility in the simultaneous characterisation of thousands of proteins in a high-throughput manner, employing a minimal amount of biological samples. These biochips can be classified as different types depending on the purpose of the microarray (i.e. analytical and functional) or their format (i.e. planar and bead-based arrays)10–12.
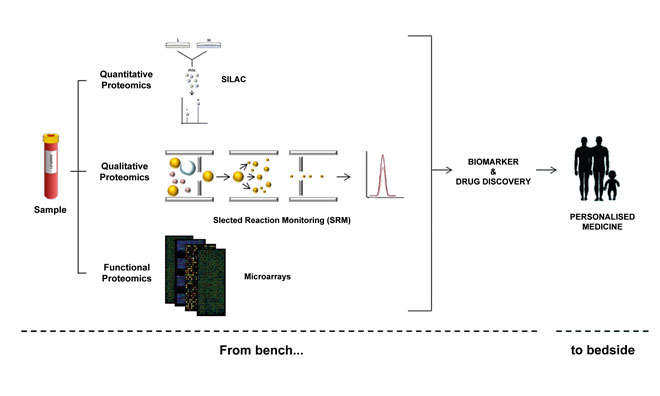
Figure 1: Protein characterisation efforts begin with quantitative, qualitative and functional strategies, with the ultimate goal being to help generate more personalised treatments
In terms of the array format, planar assays allow thousands of spots to be deposited (by contact and non-contact strategies), which contain the samples of interest in a two-dimensional surface that is chemically treated to favour the molecule anchoring2. Bead-based arrays are characterised by the deposition of the sample (i.e. antibody or protein) in a spherical surface whose diameter typically varies from 0.02 to 0.6μm. Also, they can be coloured with fluorescence molecules to produce bar-coded microspheres permitting multiple analyte testing13.
According to the samples deposited onto the surface, it is possible to distinguish protein microarrays, antibody microarrays, reverse-phase protein microarrays, tissue microarrays and DNA-based protein microarrays (e.g., nucleic acid programmable protein arrays [NAPPA]; protein in situ arrays [PISA]; DNA array to protein array [DAPA]. Also, a huge variety of printing methods has been developed to accomplish better results14,15. In this sense, contact printers are the most extended and employed due to their wide use in DNA microarrays. Briefly, contact pins are loaded with the sample to print and tightly touch onto the surface to deposit the sample (nanoliters range) in a particular spatial distribution. On the other hand, inkjet microarray printers allow samples to be deposited (picoliters range) in a highly reproducible way, producing high-quality microarray slides.
MS assays have increasingly become the most used method for analysing complex proteins in biological samples. In fact, substantial progress has been achieved in MS-based strategies thanks to its rapidly advancing, multifaceted profile and open-ended endeavours. MS can be applied to protein profiling, quantification and analysis of protein interactions and PTMs16. To this end, multiple strategies and combinations thereof are being developed for the analysis of thousands of proteins in a single sample. Thus, for quantitative proteomics label-free assays, together with relative and absolute quantification methods using stable isotope labels, are the broadest strategies performed.
Although label-free quantification is less costly and time-consuming, it is also less precise as the quantification is based on the comparison of the MS spectra to determine the protein abundance. Therefore, the usage of labels notably increases the quantification precision. Several examples of relative quantification include isotope-coded affinity tags (ICAT); tandem mass tags (TMT); isobaric tags for relative and absolute quantification (iTRAQ); and stable isotope labelling with amino acids in cell culture (SILAC). In the same way, selected reaction monitoring (SRM) is performed for absolute quantification17.
The availability of multiple methods of analysis makes it necessary to learn which one can be meaningfully applied to each particular study or biological sample of interest. Additionally, the huge amount of generated data has exponentially increased, requiring not only suitable analysis software but also public repositories for data storage and further analysis18.
Proteomics in biomarkers and drug discovery
The rapid rise of proteomics for the purpose of improving tools for protein characterisation has allowed it to be applied in the biomarker field and drug discovery. Furthermore, analysing diverse human proximal fluids (e.g. urine, serum and peripheral blood) by proteomics strategies allows cellular behaviour to be characterised by means of protein profiling. Thus, both healthy and diseased cells can be examined to map protein interactions leading to the activation of specific pathways and discovery of biomarkers of great relevance in disease diagnosis, treatment and prognosis. Another important point in this context is the role of genomics and transcriptomics, whose data provides a wealth of information that can be combined with proteomics results giving a comprehensive view of the cellular state. This integration has been recently named as proteogenomics.
The discovery of biomarkers presents notorious applications in cancer diagnosis and prognosis, including the examination of cancer heterogeneity and early detection. In fact, highly sensitive and specific biomarkers are needed in early cancer detection because tumours present high heterogeneity at protein levels and have differing protein versions, which also depend on biospecimen type and preanalytic variables. Thus, proteomics has potential in helping to design and develop more sensitive and specific detection approaches for early cancer diagnosis19,20. For instance, Hu and colleagues21 explored the human saliva proteome, looking for biomarkers targeting oral squamous cell carcinoma. A shotgun proteomics approach based on capillary reversed-phase liquid chromatography with quadrupole time-of-flight MS was employed, resulting in the detection of five candidate biomarkers.
In turn, Wood and collaborators22 reviewed the usage of urine – another noninvasive body fluid –for the early diagnosis, prognosis and monitoring of prostate, bladder and kidney cancers. Also, Ortea et al.23 have studied the potential serum biomarkers detected in rheumatoid arthritis that are related to the response to specific drugs. An iTRAQ approach was selected for the biomarker evaluation, detecting nine putative biomarkers out of 264 identified proteins between the drug responders and drug non-responders. These studies constitute some examples of the potential of proteomics for biomarker discovery.
Understanding how drugs act in the context of the cell proteome is also a task of proteomics (including clinical, chemical and functional proteomics). The drug screening is performed at drug activity, drug-target interaction and drug action mechanism levels. In this sense, clinical proteomics evaluates the efficacy, resistance and toxicity of drugs, whereas chemical proteomics estimates the specificity and selectivity of drugs, and functional proteomics maps the signalling pathways involved in drug treatments. All of them look for the discovery and broad characterisation of drugs24. For instance, a study about multi-drug resistance in Escherichia coli performed a comparative proteomics strategy (2-DE and LC-MALDI-TOF MS) to evaluate the regulation of protein expression in multi-resistant bacteria. The results concluded that proteins involved in quorum-sensing echanisms, such as S-ribosylhomocysteinelyase, could be targets for the development of new antibiotic drugs25. Also, functional protein microarrays can be applied for the identification of new targets for drug treatment by identifying differential signalling pathways26.
Diagnostic tools from proteomics
The tremendous advances in proteome characterisation have allowed numerous diseases to be profiled at protein level that, in combination with previous data and bioinformatics tools, have led to an increase of knowledge in the field. This progress is extremely significant in those cases where any previous methodology has been successful. Thus, characterising the membrane proteins is one of the major challenges for proteomics nowadays.
Of particular note is CD19, a surface protein involved in the development of many haematological disorders. Establishing the expression profiles and possible modifications present in this molecule are of great interest to understanding how these diseases evolve. Moreover, detecting the soluble CD19 levels in specific fluids could also be an interesting biomarker for disease dissemination27,28. To this end, the company ImmunoStep (www.immunostep.com) markets a kit based on microbeads for the capturing of soluble CD19 (hsCD19) presented in cerebrospinal fluid (CSF) by flow cytometry as a biomarker of CSF lymphomas. Moreover, the rapid development of proteomics strategies, as mentioned above, has led to the arising of new systems for the rapid detection of metabolites, such as the MALDI Biotyper Systems for microorganism characterisation. This tool provides high-speed, high-confidence identification and taxonomical classification of bacteria, yeast and fungi by using a MALDI-TOF MS strategy. It presents a broad application range and can be applied in environmental and pharmaceutical analysis, clinical microbiology and quality control of foods and beverages, among other uses.
Acknowledgements
The authors gratefully acknowledge financial support from the Spanish Health Institute Carlos III (ISCIII) for the grants: FIS PI114/01538. They also acknowledge Fondos FEDER (EU) and Junta Castilla-León (grant BIO/SA07/15). Fundación Solórzano FS23/2015. The Proteomics Unit belongs to ProteoRed, PRB2-ISCIII, supported by grant PT13/0001, of the PE I+D+I 2013-2016, funded by ISCIII and FEDER. P.D. is supported by a JCYL-EDU/346/2013 PhD scholarship.
 PAULA DÍEZ has a BSc in Biotechnology and an MSc degree in Cancer Biology from the University of Salamanca, Spain. She is currently doing her PhD at the Cancer Research Center in Salamanca. Her study is focused on the characterisation of B lymphocytes at the protein level by using mass spectrometry approaches and protein microarray technologies to determine the role of specific proteins and cellular signalling pathways in the development of haematological tumours. Her research interests also include the integration of different –omics strategies.
PAULA DÍEZ has a BSc in Biotechnology and an MSc degree in Cancer Biology from the University of Salamanca, Spain. She is currently doing her PhD at the Cancer Research Center in Salamanca. Her study is focused on the characterisation of B lymphocytes at the protein level by using mass spectrometry approaches and protein microarray technologies to determine the role of specific proteins and cellular signalling pathways in the development of haematological tumours. Her research interests also include the integration of different –omics strategies.
 MANUEL FUENTES graduated in chemistry and biochemistry by the University of Salamanca, Spain. After his Master’s in biotechnology at University of Bielefeld, Germany, he did a PhD at the Biocatalysis Department of the National Spanish Research Council, Madrid, Spain, which was entitled ‘Design and development of conjugation and immobilization methods of biomolecules for diagnostic methods useful in Genomics and Proteomics’. Afterwards, he moved to Harvard Institute of Proteomics at Harvard Medical School (Boston, EE.UU.) In 2010, he joined Cancer Research Center at University of Salamanca as a Scientist and as a Group Leader of the proteomics facility, focusing on biomarker and drug discovery in haematological diseases. Manuel is co-author of 95 peer-reviewed papers, nine licensed international patents, ten book chapters and over 50 invited lectures in National and international meetings.
MANUEL FUENTES graduated in chemistry and biochemistry by the University of Salamanca, Spain. After his Master’s in biotechnology at University of Bielefeld, Germany, he did a PhD at the Biocatalysis Department of the National Spanish Research Council, Madrid, Spain, which was entitled ‘Design and development of conjugation and immobilization methods of biomolecules for diagnostic methods useful in Genomics and Proteomics’. Afterwards, he moved to Harvard Institute of Proteomics at Harvard Medical School (Boston, EE.UU.) In 2010, he joined Cancer Research Center at University of Salamanca as a Scientist and as a Group Leader of the proteomics facility, focusing on biomarker and drug discovery in haematological diseases. Manuel is co-author of 95 peer-reviewed papers, nine licensed international patents, ten book chapters and over 50 invited lectures in National and international meetings.
References
- He QY, Chiu JF. Proteomics in biomarker discovery and drug development. J Cell Biochem.2003;89(5):868–86
- Gonzalez-Gonzalez M, Jara-Acevedo R, Matarraz S, Jara-Acevedo M, Paradinas S, Sayagües JM, et al. Nanotechniques in proteomics: Protein microarrays and novel detection platforms. Eur J Pharm Sci. 2012;45(4):499–506
- Díez P, González-González M, Lourido L, Dégano R, Ibarrola N, Casado-Vela J, et al. NAPPA as a Real New Method for Protein Microarray Generation. Microarrays. 2015;4(2):214–27
- Omenn GS, Lane L, Lundberg EK, Beavis RC, Overall CM, Deutsch EW. Metrics for the Human Proteome Project 2016: Progress on Identifying and Characterizing the Human Proteome, Including Post-Translational Modifications. J Proteome Res. 2016
- Paik Y-K, Jeong S-K, Omenn GS, Uhlen M, Hanash S, Cho SY, et al. The Chromosome-Centric Human Proteome Project for cataloging proteins encoded in the genome. Nat Biotechnol. 2012;30(3):221–3
- Hühmer AFR, Paulus A, Martin LB, Millis K, Agreste T, Saba J, et al. The chromosome-centric human proteome project: A call to action. J Proteome Res. 2013; 12(1):28–32
- Díez P, Droste C, Dégano RM, Gonzalez-Munoz M, Ibarrola N, Perez-Andres M, et al. Integration of Proteomics and Transcriptomics Data Sets for the Analysis of a Lymphoma B-Cell Line in the Context of the Chromosome-Centric Human Proteome Project. J Proteome Res. 2015;14(9):3530–40
- Ruiz-Romero C, Calamia V, Albar JP, Casal JI, Corrales FJ, Fernández-Puente P, et al. The Spanish biology/disease initiative within the human proteome project: Application to rheumatic diseases. J Proteomics. 2015; 127(Pt B):406–13
- Van Eyk JE, Corrales FJ, Aebersold R, Cerciello F, Deutsch EW, Roncada P, et al. Highlights of the biology and disease-driven human proteome project, 2015-2016. J Proteome Res. 2016
- Sutandy FXR, Qian J, Chen CS, Zhu H. Overview of protein microarrays. Curr Protoc Protein Sci. 2013; Chapter 27: Unit 27.1
- Dasilva N, Díez P, Gonzalez-Gonzalez M, Matarraz S, Sayagues JM, Orfao A, et al. Proteinmicroarrays: technological aspects, applications, and intellectual property. Recent Pat Biotechnol. 2013;7(2):142–52
- Lourido L, Díez P, Dasilva N, González-González M, Ruiz-Romero C, Blanco F, et al. Protein microarrays: overview, applications, and challenges. 2014; Chapter 8. Genomics and Proteomics for clinical discovery and development. Springer
- Casado-Vela J, González-González M, Matarraz S, Martínez-Esteso MJ, Vilella M, Sayagues JM. Protein arrays: recent achievements and their application to study the human proteome. Curr Prot. 2013;10(2):83-97
- Romanov V, Davidoff SN, Miles AR, Grainger DW, Gale BK, Brooks BD. A critical comparison of protein microarray fabrication technologies. Analyst. 2014;139(6):1303–26
- Fuentes M, Díez P, Casado-Vela J. Nanotechnology in the fabrication of protein microarrays. 2016; Chapter 14. Microarray technology: methods and applications, methods in molecular biology. Springer
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422(6928):198–207
- Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: A critical review. Anal Bioanal Chem. 2007;389(4):1017–31
- Riffle M, Eng JK. Proteomics data repositories. Proteomics. 2009; 9:4653–63
- Meric-Bernstam F, Akcakanat A, Chen H, Sahin A, Tarco E, Carkaci S, et al. Influence of biospecimen variables on proteomic biomarkers in breast cancer. Clin Cancer Res. 2014;20(14):3870–83
- Sallam RM. Proteomics in cancer biomarkers discovery: Challenges and applications. DisMarkers. 2015; 2015:321370
- Hu S, Arellano M, Boontheung P, Wang J, Zhou H, Jiang J, et al. Salivary proteomics for oral cancer biomarker discovery. Clin Cancer Res. 2008;14(19):6246–52
- Wood SL, Knowles M a, Thompson D, Selby PJ, Banks RE. Proteomic studies of urinarybiomarkers for prostate, bladder and kidney cancers. Nat Rev Urol. 2013;10(4):206–18
- Ortea I, Roschitzki B, López-Rodríguez R, Tomero EG, Ovalles JG, López-Longo J, et al. Independent candidate serum protein biomarkers of response to adalimumab and to infliximab in rheumatoid arthritis: An exploratory study. PLoS One. 2016;11(4):e0153140
- Savino R, Paduano S, Preianò M, Terracciano R. The proteomics big challenge for biomarkers and new drug-targets discovery. Int J Mol Sci. 2012;13(11):13926–48
- Piras C, Soggiu A, Bonizzi L, Gaviraghi A, Deriu F, De Martino L, et al. Comparative proteomics to evaluate multi drug resistance in Escherichia coli. Mol Biosyst. 2012;8(4):1060-7
- Díez P, Lorenzo S, Dégano RM, Ibarrola N, González-González M, Nieto W. Multipronged functional proteomics approaches for the global identification of altered cell signalling pathways in B-cell chronic lymphocytic leukemia. Proteomics. 2016; 16, 1193-1203
- Muñiz C, Martín-Martín L, López A, Sánchez-González B, Salar A, Almeida J, et al. Contribution of cerebrospinal fluid sCD19 levels to the detection of CNS lymphoma and its impact on disease outcome. Blood. 2014;123(12):1864–9
- Galicia N, Díez P, Dégano RM, Ibarrola N, Fuentes M. Biomarker identification in CSF for leptomeningeal metastases by proteomics approaches. 2016; Chapter. Proteomics Methods in Neuropsychiatric Research (in press)
Related topics
Proteomics



nice post