Patient-derived neural progenitor cells as a drug discovery model for mitochondrial diseases
Posted: 13 December 2017 | Alessandro Prigione, Carmen Lorenz, Gizem Inak | No comments yet
Mitochondrial DNA (mtDNA) mutations cause severe disorders that are untreatable and mostly affect the nervous system. The difficulty in funding therapies may also be explained by the lack of viable modelling systems…
Animal models are scarce due to the challenge of mtDNA engineering and cell types used for compound screening are mostly limited to peripheral cells from patients or immortalised cell lines. However, these cells do not share the same functional and metabolic features of the nervous system cells that are affected in patients. We recently showed that neural progenitor cells (NPCs) differentiated from patient-derived induced pluripotent stem cells (iPSCs) represent an effective drug discovery cell model for mtDNA disorders. We believe that their use may pave the way for the identification of novel therapeutics against these hitherto incurable human diseases.
The major task of mitochondria is to produce energy in the form of ATP, which is used by every cell. The functionality of the mitochondria is under the control of both the nuclear and the mitochondrial genome. Mitochondrial DNA (mtDNA) encodes for only 37 genes encoding for proteins of the electron transport chain (ETC) and ATP synthase located in the inner mitochondrial membrane. mtDNA is exclusively maternally inherited and is present in multiple copies within each cell. This latter feature is very important for understanding the pathogenesis of mtDNA mutations. In fact, a certain mutation can be either homoplasmic, when it is present in all the mtDNA copies, or heteroplasmic, when it is present in only some of the mtDNA copies. The level of heteroplasmy can associate with the biological effect of the mutation.
Mutations in mtDNA occur with a minimum disease prevalence of one per 5,000.1 Deletions or point mutations have been reported in virtually every mtDNA gene. The mutations are strongly linked to diseases affecting the nervous system with various clinical phenotypes ranging in severity from asymptomatic to fatal within the first years of life.2,3 Importantly, no effective treatment exists for mtDNA diseases.
One of the major hurdles for the discovery of therapies is the paucity of model systems available. In fact, it has been difficult to develop animal models for mitochondrial disease, due to challenges inherent in engineering mtDNA. Existing cellular models lack the functional and metabolic features of neural cells. Moreover, they do not provide the patient-specific match between mitochondrial and nuclear genomes. This aspect is crucial, since specific characteristics of an individual nuclear DNA may influence the course of these diseases.
Using patient-derived neural cells in drug discovery
A decade ago, Shinya Yamanaka demonstrated that adult somatic cells can be reprogrammed into embryonic-like cells.4,5 The resulting cells, dubbed induced pluripotent stem cells (iPSCs), share the same properties as embryonic stem cells (ESCs). In addition, iPSCs can be derived from every individual from easily accessible peripheral cells. Hence, iPSCs do not have any ethical limitations. More importantly, iPSCs retain the ability to differentiate into cell types of the body, including non-accessible cells like those belonging to the central nervous system.
With the progress of iPSC technology, modelling of complex neurological diseases is now possible. Patient-derived cells can be reprogrammed to iPSCs and further differentiated into neuronal cells. Using this approach, several kinds of human neuronal subtypes can be obtained in a dish, including midbrain dopaminergic neurons,6,7,8 GABAergic neurons,9,10 and serotonergic neurons.11
Unfortunately, however, neuronal differentiation from iPSCs is cumbersome, costly, and extremely time-consuming. Complex protocols are necessary to reach neuronal maturation and appropriate expression of protein markers. Furthermore, the obtained cultures are mostly heterogeneous with batch-dependent differences.12,13,14 Therefore, several hurdles need to be overcome to allow the use of iPSC-derived neurons in high-throughput (HT) studies. In fact, HT drug discovery platforms require the reproducibility and robustness of the cell model used. Targeting mitochondrial diseases with iPSC-derived NPCs In order to advance our understanding of the pathogenetic mechanisms of mitochondrial disorders, it is therefore critical to establish appropriate model systems. These systems should fulfil three main properties:
- Allow reproducible results amenable to HT experiments
- Recapitulate the functional and metabolic features of the patient neural cells
- Maintain the patient-specific nuclear and mitochondrial genome.
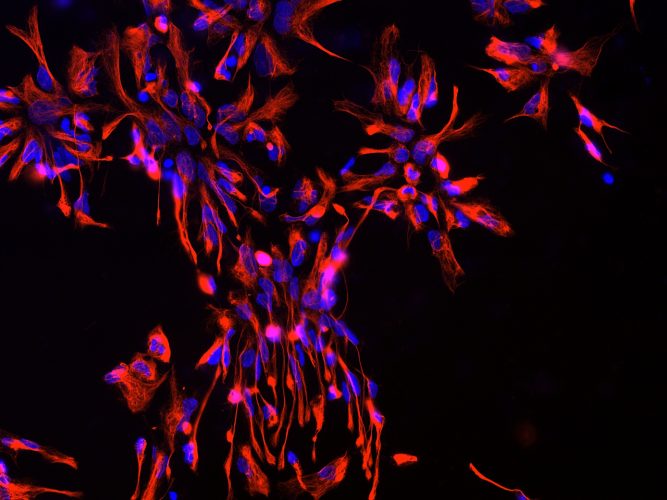
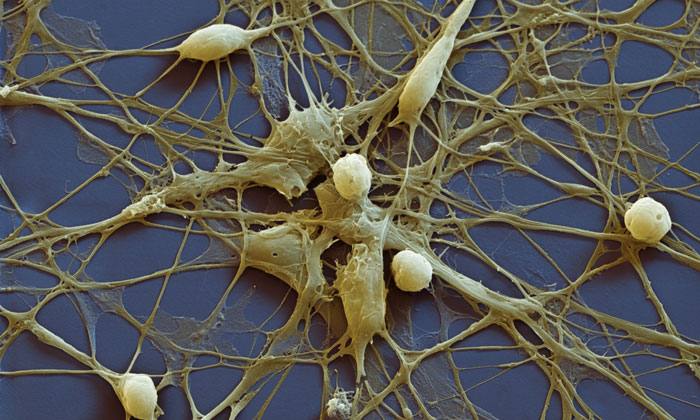
Figurer 1: Neural progenitor cells (NPCs) derived from iPSCs. Blue: nuclear staining with Hoechst; Red: NPC marker NESTIN.
Many of the differentiation protocols used for neuronal generation from iPSCs require the intermediate induction of neural progenitor cells (NPCs). NPCs are multipotent cells that can give rise to both neurons and glia (Figure 1). In vivo, NPCs enable neurogenesis even in the adult human brain15,16 and are located in defined brain regions, ie, the subgranular zone of the dentate gyrus in the hippocampus and the subventricular zone of the cortex.17,18 In vitro, NPCs can be rapidly produced from iPSCs using different strategies, including the formation of embryoid bodies (EBs) followed by manual isolation of NPCs from neural rosettes.19,20 As a rapid alternative, small molecules can be applied to convert iPSCs directly into NPCs in only one week.21,22 Importantly, our recent data showed that the transcriptomic iPSC-derived NPCs are similar to that of brain-derived NPCs.23
In comparison to fully mature neurons, NPCs may hold key advantages for drug discovery applications.24 First, NPCs can be readily obtained from iPSCs in a cost-effective fashion. Second, the resulting cell population is mainly homogeneous and does not require additional purification steps. Third, NPCs are proliferative, which makes their cultivation and expansion relatively feasible.
In order to employ NPCs for mitochondrial diseases, it is important to determine whether the cells rely on functional mitochondrial metabolism. We and others demonstrated that during the generation of NPCs from human and mouse PSCs, mitochondria undergo a functional maturation.23,25 This maturation leads to elongated organelle morphology with clearly defined cristae. In parallel, there is an increase in mitochondrial mass and mtDNA content.26 This remodelling of mitochondrial morphology during NPC derivation is associated with a metabolic shift towards oxidative phosphorylation (OXPHOS)- based metabolism, while glycolysis and lactate production are reduced.23 These data imply that NPCs exhibit a metabolic state that depends on mitochondrial functionality.
In our recent work, we demonstrated that during generation of patient-derived iPSCs and subsequent neural induction to NPCs, the mitochondrial genome is entirely retained.23 At the same time, no additional mtDNA modifications were observed. This aspect is crucial as it can allow us to assume that every kind of phenotype observed in the culture is only due to the patient-specific mutations and not to reprogramming-induced artefacts.
In fact, given that NPCs show increased OXPHOS metabolism, it was possible that the consequent increase in reactive oxygen species (ROS) production may damage the mitochondrial genome.27 However, this was not the case. This is very important, as the failure to maintain mtDNA integrity can lead to impaired neuronal maturation.28
Compound screenings for mitochondrial diseases using patient NPCs
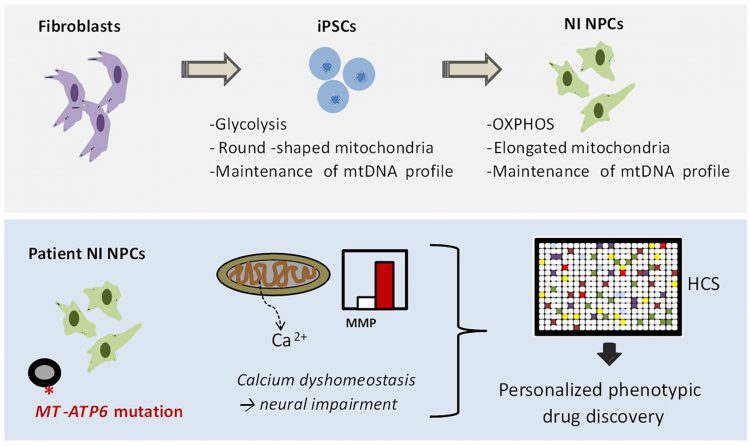
FIGURE 2: NPC-based drug discovery for mitochondrial disorders. In our recent manuscript,23 we first demonstrated that iPSC-derived NPCs exhibit a metabolism dependent on mitochondrial oxidative phosphorylation (OXPHOS) and retain the mtDNA profile of the patient somatic cells. We next generated iPSC-derived NPCs generated from patients carrying the mutation m.9185T>C in the mitochondrial gene MT-ATP6. We identified increased mitochondrial membrane potential (MMP) and calcium defects in patient NPCs as potential causes for neural impairment. Finally, we employed these phenotypes to develop a high-content screening (HCS) method for screening FDA-approved drugs directly on patient NPCs.
In our recent work, we demonstrated that iPSC-derived NPCs exhibit the correct functional, metabolic, and genetic features to be used for drug discovery of mitochondrial diseases (Figure 2).
As a proof-of-concept, we generated NPCs from patients carrying a homoplasmic mutation in the mitochondrial gene MT-ATP6 (m. 9185T>C), which is a known pathogenetic cause of Leigh syndrome.29 We observed that patient NPCs show defects in ATP production and abnormal increase in the mitochondrial membrane potential (MMP). Furthermore, patient NPCs exhibited impaired mitochondrial calcium homeostasis, which might represent a potential cause of neural impairment.
Having identified these patient-specific cellular phenotypes, we employed patient NPCs to carry out a small-scale high-content screening (HCS) using FDA-approved drugs. The screening aimed to identify compounds capable of ameliorating the abnormally increased MMP of patient NPCs. We discovered that one compound was able to do that: the phosphodiesterase type 5 (PDE5) inhibitor avanafil. By applying avanafil overnight on patient NPCs and patient neurons, we were then able to partially rescue the mitochondrial calcium defects. PDE5 inhibitors are FDA-approved drugs and are already employed safely in some paediatric conditions (for example, pulmonary hypertension). Hence, their repositioning in the context of Leigh syndrome may potentially allow the quick translation of our findings into medical applications.
Taken together, NPCs allow phenotypic drug discovery for mitochondrial disorders affecting the nervous system. This promising iPSC-based strategy provides a useful in vitro platform for a better understanding of mechanisms in mitochondrial disorders, and for the identification of therapeutic approaches for these hitherto incurable human diseases.
Acknowledgements
We declare no competing financial or commercial interests and acknowledge financial support from the Bundesministerium für Bildung und Forschung (BMBF) (e:Bio Young Investigator grant AZ. 031A318) and the Berlin Institute of Health (BIH).
References
- Gorman GS, Schaefer AM, Ng Y et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015;77:753–759. DOI: 10.1002/ ana.24362
- Schon EA, DiMauro S, Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat Rev Genet. 2012;13:878-890. DOI: 10.1038/nrg3275
- Lieber DS, Calvo SE, Shanahan K et al. Targeted exome sequencing of suspected mitochondrial disorders. Neurology. 2013;80:1762-1770. DOI: 10.1212/ WNL.0b013e3182918c40
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676 DOI:10.1016/j. cell.2006.07.024
- Takahashi K, Tanabe K, Ohnuki M et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861-72. DOI: 10.1016/j.cell.2007.11.019
- Chambers SM, Fasano CA, Papapetrou EP et al. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nature Biotechnology. 2009;27(3):275– 280. DOI: 10.1038/nbt.1529
- Kriks S, Shim JW, Piao J et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature. 2011;480:547–551. DOI: 10.1038/ nature10648
- Reinhardt P, Glatza M, Hemmer K et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS One.2013;8,e59252. DOI: 10.1371/journal.pone.0059252
- Maroof AM, Keros S, Tyson JA et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell 2013;12:559-572. DOI: 10.1016/j.stem.2013.04.008
- Nicholas CR, Chen J, Tang Y et al. Functional maturation of hPSC-derived forebrain interneurons requires an extended timeline and mimics human neural development. Cell Stem Cell.2013;12:573-586. DOI: 10.1016/j.stem.2013.04.005
- Erceg S, Laınez S, Ronaghi M et al. Differentiation of Human Embryonic Stem Cells to Regional Specific Neural Precursors in Chemically Defined Medium Conditions. PLoS ONE. 2008;3(5):e2122. DOI: 10.1371/journal.pone.0002122
- Boulting GL, Kiskinis E, Croft GF et al. A functionally characterized test set of human induced pluripotent stem cells. Nat Biotechnol. 2011;29:279-286. DOI: 10.1038/nbt.1783
- Nguyen HN, Byers B, Cord B et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 2011;8:267-280. DOI: 10.1016/j. stem.2011.01.013
- Di Giorgio FP, Boulting GL, Bobrowicz S and Eggan KC. Human embryonic stem cell-derived motor neurons are sensitive to the toxic effect of glial cells carrying an ALS-causing mutation. Cell Stem Cell 2008; 3, 637-648. DOI: 10.1016/j.stem.2008.09.017
- Kempermann G. New neurons for survival of the fittest. Nat Rev Neurosci. 2012;13: 727:736, DOI: 10.1038/nrn3319
- Ninkovic J, Götz M. How to make neurons–thoughts on the molecular logic of neurogenesis in the central nervous system. Cell Tissue Res. 2015;3595-16, DOI: 10.1007/s00441-014-2048-9
- Alvarez-Buylla A, Temple S. Stem cells in the developing and adult nervous system. J Neurobiol. 1998;36:105–110. PMID: 9712298
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660, DOI: 10.1016/j.cell.2008.01.033
- Elkabetz Y, Panagiotakos G, Shamy G et al. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes & Development. 2008;22:152-165. DOI: 10.1101/gad.1616208
- Koch P, Opitz T, Steinbeck JA et al. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc Natl Acad Sci USA. 2009;106:3225-3230. DOI: 10.1073/pnas.0808387106
- Li W, Sun W, Zhang Y et al. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc Natl Acad Sci USA. 2011;108(20):8299-304. DOI: 10.1073/pnas.1014041108
- Reinhardt P, Glatza M, Hemmer K et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS One. 2013;8,e59252. DOI: 10.1371/journal.pone.0059252
- Lorenz C, Lesimple P, Bukowiecki R, Zink A, Inak G, Prigione A. Human iPSC-derived neuronal progenitors are an effective drug discovery model for neurological mitochondrial DNA disorders. Cell Stem Cell. 2017;20(5):659-674. DOI: 10.1016/j.stem.2016.12.013
- Inak G, Lorenz C, Lisowski P, Zink A, Mlody B, Prigione A. Concise Review: Induced Pluripotent Stem Cell-Based Drug Discovery for Mitochondrial Disease. Stem Cells. 2017; [Epub ahead of print] DOI: 10.1002/stem.2637
- Choi HW, Kim JH, Chung MK et al. Mitochondrial and metabolic remodeling during reprogramming and diff erentiation of the reprogrammed cells. Stem Cells Dev. 2015;24:1366– 1373 DOI: 10.1089/scd.2014.0561
- Cho YM, Kwon S, Pak YK et al. Dynamic changes in mitochondrial biogenesis and antioxidant enzymes during the spontaneous diff erentiation of human embryonic stem cells. Biochem Biophys Res Commun. 2006;348:1472–1478, DOI: 10.1016/j.bbrc.2006.08.020
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging, Cell. 2005;120:483-495. DOI: 10.1016/j.cell.2005.02.001
- Wang W, Osenbroch P, Skinnes R et al. Mitochondrial DNA integrity is essential for mitochondrial maturation during differentiation of neural stem cells. Stem Cells. 2010;28:2195-2204, DOI: 10.1002/stem.542
- Auré K, Jardel C, Clarysse L et al. Episodic weakness due to mitochondrial DNA MT-ATP6/8 mutations. American Academy of Neurology. 2013;81:1810-1818. DOI: 10.1212/01. wnl.0000436067.43384.0
Biography
 DR ALESSANDRO PRIGIONE* is Independent Team Leader at the Max Delbrück Center for Molecular Medicine (MDC) in Berlin. He received an MD from the University of Milan in 2002 and a PhD from the University of Milan-Bicocca in 2008. During his PhD, Dr Prigione worked for two years at the University of California in Davis. He carried out his postdoctoral work at the San Raffaele Institute in Milan and at the Max Planck Institute for Molecular Genetics in Berlin.
DR ALESSANDRO PRIGIONE* is Independent Team Leader at the Max Delbrück Center for Molecular Medicine (MDC) in Berlin. He received an MD from the University of Milan in 2002 and a PhD from the University of Milan-Bicocca in 2008. During his PhD, Dr Prigione worked for two years at the University of California in Davis. He carried out his postdoctoral work at the San Raffaele Institute in Milan and at the Max Planck Institute for Molecular Genetics in Berlin.
* CORRESPONDING AUTHOR Alessandro Prigione, MD, PhD. Robert-Roessle-Str. 10, 13125 Berlin, Germany. Phone: 0049-30-9406- 2871. Fax: 0049-30-9406- 3696. Email: alessandro. [email protected].
 CARMEN LORENZ is a PhD student in her fourth year in the team of Dr Prigione at the MDC in Berlin. Her project deals with the development of iPSC-based strategies for studying maternally inherited Leigh syndrome. The project is supported by a PhD Translational Fellowship from the Berlin Institute of Health (BIH).
CARMEN LORENZ is a PhD student in her fourth year in the team of Dr Prigione at the MDC in Berlin. Her project deals with the development of iPSC-based strategies for studying maternally inherited Leigh syndrome. The project is supported by a PhD Translational Fellowship from the Berlin Institute of Health (BIH).
 GIZEM INAK is currently doing her PhD in Dr Prigione’s Lab at the (MDC) in Berlin. She completed her Master’s degree in Cellular and Molecular Neuroscience in Tuebingen. Her PhD project is focusing on functional and bioenergetic defects in iPSC-derived dopaminergic neurons from patients with Leigh Syndrome.
GIZEM INAK is currently doing her PhD in Dr Prigione’s Lab at the (MDC) in Berlin. She completed her Master’s degree in Cellular and Molecular Neuroscience in Tuebingen. Her PhD project is focusing on functional and bioenergetic defects in iPSC-derived dopaminergic neurons from patients with Leigh Syndrome.
Related topics
Disease Research, Drug Discovery, Genomics, Metabolomics, Neurosciences, Screening
Related conditions
Leigh syndrome
Related organisations
Berlin Institute of Health (BIH), Max Delbrück Centre for Molecular Medicine
Related people
Alessandro Prigione, Carmen Lorenz, Gizem Inak