Should LC-MS/MS proteomics guide targeted drug selection for cancer patients?
Posted: 22 September 2015 | David Britton (Barts Cancer Institute), Pedro Cutillas (Barts Cancer Institute) | No comments yet
History has demonstrated that the use of companion in vitro diagnostic tests to select cancer patients positive for a drug target significantly improves response to targeted anti-cancer therapies…
Many such therapies (74 in total) have been approved by the US Food and Drug Administration and this number will continue to increase, as well as the number of cancer types they will be prescribed for. Consequently, as therapeutic options increase, methods that measure one or just a few biomarkers at a time have limitations as companion diagnostics. There is therefore a need to apply quantitative tumour molecular profiling technologies to select the drugs more likely to produce a response in each individual patient. Here we discuss the rationale of using LC-MS/MS proteomics as a companion diagnostic tool to identify the best drug target in each individual cancer patient.
The number of cancer patients that survive the disease is now larger than ever before1. In addition to improved surgery, better prevention and early detection methods, the development of targeted anti-cancer drugs is contributing to this partial success story of modern medicine2,3. However, cancer is a very heterogeneous disease; even tumours from the same tissue of origin show a wide range molecular and biochemical features that distinguish them from one another. The practical consequence of this heterogeneity is that not all patients respond to treatment equally well. Indeed, intense research on the molecular causes of cancer have resulted in the development of therapies that rationally target the biochemical aberrations that drive the malignant phenotype, yet most patients fail to benefit from this new set of targeted therapies2,3. A molecular and biochemical understanding as to why some patients respond to new cancer drugs, while others do not thrive on the same treatments, is of paramount importance for designing therapies personalised to each individual cancer patient.
Hundreds of targeted compounds are being tested to treat different forms of cancer. Progression of these new compounds into drugs of clinical utility requires selecting the tumour subtype that will more likely benefit from treatment2,3. In addition, oncologists already have several therapeutic options for a few cancer types; with the approval of new compounds currently in trials, these options will significantly increase in the not so distant future. Thus oncologists often face the question of which drug, out of the many available, should a given patient receive, and this dilemma will be more acute as new drugs go through regulatory approval.
In this article we consider the role that proteomics methodology will have in selecting the cancer drug (or drug combination) that will more likely produce a therapeutic response in each individual cancer patient. The pace of advance in proteomic and bioinformatics technology is such that it may soon be possible to fully characterise tumour biochemistry with the depth and throughput required to accurately rationalise the drugs that should produce a stronger therapeutic response in each individual patient.
Development of personalised cancer treatments
The concept of personalised medicine is not new. In the late 19th century Sir George Beatson recognised a link between the ovaries and the neoplastic breast4. By the 1970s, oestrogen binding protein, known now as oestrogen receptor (ER), had been identified, as well as the relationship between ER status of breast tumours and response to antioestrogen therapy, with ER-positive patients responding significantly better than those who were ER-negative5,6,7,8. This relationship prompted the routine testing of breast cancer patient tumour biopsies for ER in order to predict response to antioestrogen therapy and thereby determine treatment strategy5,6,7,8.
The Herceptin® (trastuzumab) story is another classic case of a protein drug target being quantified in primary tumour tissue to allow patient responses to be predicted and their selection for targeted therapy with trastuzumab, which is a monoclonal antibody targeting HER29. HER2 is also used as a prognostic marker and is predictive for other systemic therapies such as tamoxifen and chemotherapeutic agents9. In 2000, after recognising the importance of HER2 screening, the ASCO Tumour Marker Guidelines Panel recommended routine immunohistochemical testing of HER2 expression on newly-diagnosed and metastatic breast cancer, which was then formally standardised in 2006 by ASCO and the College of American Pathologists (CAP), and further updated in 20139,10,11.
The use of companion in-vitro diagnostic (IVD) tests to pre-select patients who are most likely suitable for antioestrogen or anti-HER2 therapies has proven to be clinically beneficial by significantly increasing response rates. Indeed, without patient selection based on HER2 protein expression, trastuzumab clinical trials would not have been statistically significant and the drug would have not been approved for therapy9,12,13,14. This highlights the advantage of directly measuring expression of protein drug targets, both for suitability/selection for clinical trials and for the routine personalisation of patient treatment.
Resistance problems
Despite their partial success, intrinsic and acquired resistance to targeted therapy limits their utility2,3,15. Clearly, the most common cause of resistance to a targeted drug is absence of its protein target in the tumour. However patients with tumours classified as positive for a protein drug target/predictive marker may still experience resistance to therapy. This phenomenon is highlighted by the fact that 30% of ER-positive breast cancers are intrinsically resistant to antiestrogenic agents, and 50% of patients that initially respond eventually relapse16. Similarly, nearly 70% of HER2-positive patients with metastatic breast cancer possess intrinsic resistance to anti-HER2 therapies and nearly all develop resistance after initial responsiveness17,18. Thus expression of the target is not the only reason why patients respond to targeted therapies.
A predominant cause of resistance to antiestrogen and anti-HER2 therapy is signalling through alternative, downstream or parallel pathways15,16,17,18. Indeed, signalling from receptor tyrosine kinases (RTKs) such as EGFR/HER2 and downstream RAS/RAF/MEK/ERK or PI3K/AKT/mTOR signalling is associated with tamoxifen resistance, particularly in Luminal B cancers16,19. Resistance to trastuzumab has been attributed to mutations that activate intracellular signalling downstream of HER2, including mutations in PIK3CA (phosphoinositide 3-kinase class Iα) and/or loss of function of PTEN, as well as signalling through other distinct pathways (e.g. emanating from MET, IGF-1R, EphA2, or EpoR)17,18.
New targeted therapies
Kinases have provided a treasure trove of drug targets. Classic examples of aberrant kinases in cancer include the tyrosine kinase breakpoint cluster region-Abelson (BCR-ABL) in chronic myelogenous leukaemia, the serine/threonine kinase BRAF in melanoma and EGFR in lung cancer20. Many kinase inhibitors are approved or in advanced stages of clinical development, including those that target RTKs directly (e.g., EGFR, HER2, Met, KIT, FLT-3), serine/threonine kinases (e.g., BRAF, mTOR, Erk and Akt), the dual specificity protein kinase MEK, lipid kinases (several PI3K isoforms), and non-receptor tyrosine kinases (such as Src, Yes, Fyn and BCR-Abl)21,22. The number of targeted therapies will continue to grow as more drugs are developed against known drug targets, new targets are identified and approved targeted therapies are redeployed in additional cancer types. In fact, cancer diagnosis will have less emphasis on tissue/cell type of origin but more on tumour molecular profile classification that predicts best therapeutic regimes.
As is the case for resistance to antioestrogen or anti-HER2 therapies, resistance to the plethora of targeted therapies can in many cases also be attributed to oncogenic bypass or signalling redundancy and this may occur in cancer cells that are positive or negative for drug target (Figure 1). Resistance-causing pathways may already be active in a subpopulation of cancer cells within a tumour that contain the active drug target (ADT) prior to treatment (Figure 1, A.b), or activated in ADT-positive cancer cells upon treatment (Figure 1, A.c). They can alternatively be active in cancer cells that do not contain ADT (Figure 1, B.a & B.b). This intra-tumoural heterogeneity may be a result of clonal evolution or cancer stem cells, or indeed a combination of the two models whereby some of the cancer stem cells over repeated divisions experience genetic instability15,23. This can give rise to further mutations and the emergence of different populations of cancer stem cells. Each of these populations then has the potential to create sub-clones of more differentiated cancer cells which retain their proliferative capabilities, yet each utilising different signalling pathways to drive the cancer phenotype15,23.
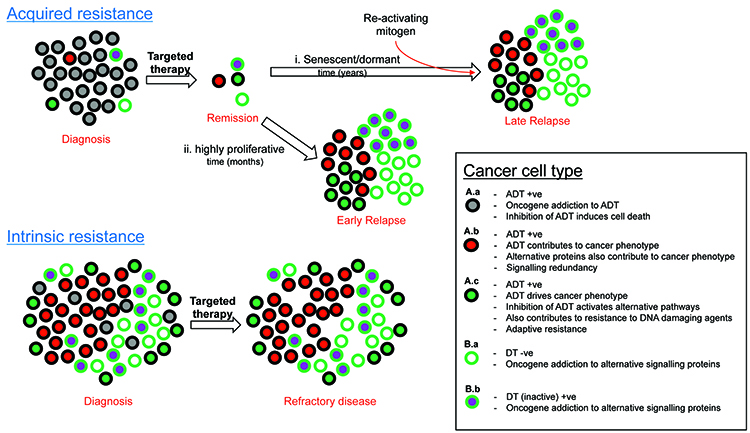
Figure 1: Categorising causes of resistance to target therapy.
The level of heterogeneity and the proportion of each cell type relative to each other may determine response or the type of resistance to therapy. In the case of solid tumours, resistance to targeted therapy is particularly prevalent in the metastatic setting, but also possible in the adjuvant setting due to undetectable micro-satellites or circulating tumour micro-emboli. Haematological cancers such as leukaemia, lymphoma and multiple myeloma may also show resistance to targeted therapy via the same principles shown in Figure 1. It is worth noting that already active (Figure 1, A.b) or activation (Figure 1, A.c) of signalling pathways may also occur during resistance to cytotoxic chemotherapies. Other important causes of resistance to consider include poor drug delivery to and into cancer cells, inappropriate drug metabolism within cancer cells and excessive efflux of drug out of cancer cells (reviewed in Holohan et al15).
LC-MSMS proteomics
Due to the myriad of potential causes of resistance to targeted therapies involving oncogenic bypass and pathway redundancy, it is difficult to always accurately predict whether a patient is likely to respond to a therapy using technologies that only measure one or a few predictive markers. In fact, since hundreds of anti-cancer drug targets and oncogenic signalling proteins are relevant to therapeutic selection (as discussed above), measuring expression and activation status of all of these using the current gold standard analysis, namely immunohistochemistry (IHC), is not feasible. Therefore a more global tumour profile at the protein level is required.
Genomic technologies are highly effective at identifying activation and loss of function mutations that might predict response; however unexpected resistance can be seen even after predicting treatment using whole genome sequencing, perhaps because genetic mutations do not provide a readout of the drug target expression and certainly offer no clues as to its activity. For example, cancer cells may contain activating BRAF mutations in their exome but this does not necessarily correlate with expression of the active enzyme. Furthermore, enzymatic activity and protein-protein interactions, the processes that targeted drugs are designed to inhibit, are frequently modulated by extracellular signals that emanate from the tumour microenvironment and which are therefore undetectable by genomic technologies. Similarly, transcriptomic data is not fully reliable at measuring drug target expression, since a linear relationship between mRNA and protein expression does not always exist, and protein amounts may or may not correlate with their enzymatic activity. Reversed-phase protein (RPPA) microarrays possess limitations due to the difficulty of obtaining a comprehensive antibody repertoire with the required specificity. Furthermore, due to differences in the epitope/antibody affinity of each antibody on the array, it is impossible to quantify abundance and activity from a single biopsy.
LC-MS/MS proteomics is proven to directly quantify protein expression of multiple drug targets from tumour tissue simultaneously in an unbiased manner24,25,26. There are many different LC-MS/MS proteomic workflows and methods, each with their strengths and weaknesses depending on the application. A particular area of excitement, however, is label-free quantification due to its ability to rank proteins in terms of abundance in a single sample. The technology is applicable to cancer cells from a single small biopsy, such as those obtained from a solid tumour or blasts from haematological cancers. The advantage of this approach is that from a single biopsy the abundance of all drug targets may be ranked from highest abundance to lowest26.
In addition to protein abundance, the technique can also be used to quantify enzymatic activity by measuring posttranslational modifications. An example is phosphoproteomics, which is the subfield of proteomics concerned with the analysis of protein phosphorylation. As each phosphorylation site is by definition the result of net kinase activity, with appropriate algorithms, net kinase activities may be inferred from the phosphoproteomic data27,28,29. Thus, with this approach (Figure 2), abundance of most drug targets, and activity of most kinases, may be ranked within the biopsied specimen, thus giving the implication of an ideal therapy for the patient.
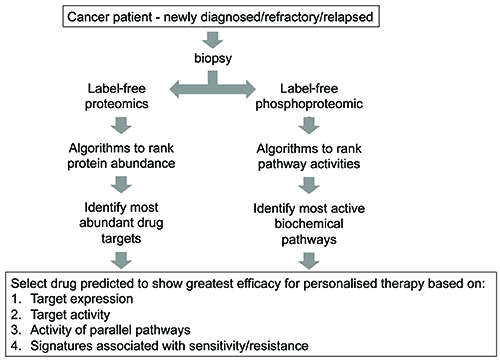
Figure 2: Proposed strategy utilising LC-MS/MS proteomics to select appropriate targeted therapy.
Despite the fact that LC-MS/MS-based proteomics provides an opportunity to improve prediction to drug responses, obstacles to be overcome remain. As with all methodologies, specimen freshness is critical to ensure good recovery of proteins and their post-translationally modified counterparts; therefore tissue should be snap frozen or heat inactivated, as soon as possible following removal from the patient. Clear regulatory guidelines must also be established to ensure standard operating procedures are of a high standard to produce reproducible and accurate results.
In summary, targeted therapies have been developed through a rational understanding of tumour biochemistry. Current methodology for label-free proteomics offers the opportunity to analyse such biochemistry with unprecedented detail and with a throughput and robustness that now make the technique suitable for clinical implementation. Therefore, proteomics data, together with new bioinformatics tools for their analysis and interpretation, should help oncologists to make even more rational decisions when trying to select the right drug for the right patient.
Biographies
 DAVID J. BRITTON, PhD, is Postdoctoral Scientist at the Bart’s Cancer Institute, Queen Mary University of London. He designs, optimises and applies LC-MS/MS proteomic workflows to identify resistance mechanisms to anti-neoplastic therapies with an aim to identify new drug targets, predict response, or guide suitable therapy selection. Prior to this, Dr. Britton worked at Proteome Sciences plc as Senior Research Scientist and before that at Thermo Scientific as an LC-MS/MS training instructor specialising in Orbitrap and ion trap instruments. He obtained a BSc in Pharmacology from the University of Bristol in 2001, then went on to do a PhD at the Tenovus Laboratories, Cardiff University and then to California to work as a postdoctoral fellow under the supervision of Profs Chris Benz and Brad Gibson at the Buck Institute between 2005 and 2008.
DAVID J. BRITTON, PhD, is Postdoctoral Scientist at the Bart’s Cancer Institute, Queen Mary University of London. He designs, optimises and applies LC-MS/MS proteomic workflows to identify resistance mechanisms to anti-neoplastic therapies with an aim to identify new drug targets, predict response, or guide suitable therapy selection. Prior to this, Dr. Britton worked at Proteome Sciences plc as Senior Research Scientist and before that at Thermo Scientific as an LC-MS/MS training instructor specialising in Orbitrap and ion trap instruments. He obtained a BSc in Pharmacology from the University of Bristol in 2001, then went on to do a PhD at the Tenovus Laboratories, Cardiff University and then to California to work as a postdoctoral fellow under the supervision of Profs Chris Benz and Brad Gibson at the Buck Institute between 2005 and 2008.
 PEDRO RODRIGUEZ CUTILLAS leads the Integrative Cell Signalling and Proteomics Group at Barts Cancer Institute in London. This group uses proteomics approaches to the investigation of cell signalling in health and disease. Dr Cutillas obtained a PhD in 2004 from University College London under the supervision of Robert Unwin, Mike Waterfield and Al Burlingame. He then worked as a postdoctoral fellow in the group of Bart Vanhaesebroeck at the Ludwig Institute for Cancer Research before obtaining a lectureship from Barts School of Medicine in 2007. He and his group developed technology based on mass spectrometry to quantify cell signalling and then used these new methods to investigate mechanism by which in cancer cells respond to therapies that target signalling enzymes.
PEDRO RODRIGUEZ CUTILLAS leads the Integrative Cell Signalling and Proteomics Group at Barts Cancer Institute in London. This group uses proteomics approaches to the investigation of cell signalling in health and disease. Dr Cutillas obtained a PhD in 2004 from University College London under the supervision of Robert Unwin, Mike Waterfield and Al Burlingame. He then worked as a postdoctoral fellow in the group of Bart Vanhaesebroeck at the Ludwig Institute for Cancer Research before obtaining a lectureship from Barts School of Medicine in 2007. He and his group developed technology based on mass spectrometry to quantify cell signalling and then used these new methods to investigate mechanism by which in cancer cells respond to therapies that target signalling enzymes.
References
- Cancer survival statistics | Cancer Research UK [Internet]. [cited 2015 Jul 14]. Available from: http://www.cancerresearchuk.org/health-professional/cancer-statistics/survival#heading-Zero
- Cho S-H, Jeon J, Kim S Il. Personalized Medicine in Breast Cancer: A Systematic Review. J Breast Cancer. 2012;15(3):265
- Huang M, Shen A, Ding J, Geng M. Molecularly targeted cancer therapy: Some lessons from the past decade. Trends Pharmacol Sci. 2014;35(1):41–50
- Beatson G. On the Treatment of Inoperable Cases of Carcinoma of the Mamma: Suggestions for a New Method of Treatment with Illustrative Cases. Lancet. 1896 Jul 11;148(3802):104–7
- Perry MC. The Chemotherapy Source Book. Wolters Kluwer Health/Lippincott Williams & Wilkins; 2008
- McGuire WL. Estrogen receptors in human breast cancer. J Clin Invest. 1973;52:73–7
- Dowsett M. Estrogen receptor: Methodology matters. J Clin Oncol. 2006;24(36):5626–8
- Wolff AC, Dowsett M. Estrogen Receptor: A never Ending Story. J Clin Oncol. 2011;29(22):2955–8
- Wolff AC, Hammond MEH, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. J Clin Oncol. 2007;25(1):118–45
- Bast R. Summary of recommendations. J Clin Oncol. 2001;19(21):4185–8
- Wolff AC, Hammond MEH, Hicks DG, Dowsett M, McShane LM, Allison KH, et al. Recommendations for human epidermal growth factor receptor 2 testing in breast cancer: American Society of Clinical Oncology/College of American Pathologists clinical practice guideline update. J Clin Oncol [Internet]. 2013;31(31):3997–4013
- Slamon D. Use of Chemotherapy Plus a Monoclonal Antibody Against Her2. N Engl J Med [Internet]. 2001;344(11):783–92
- Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehrenbacher L, et al. Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999;17(9):2639–48
- Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol. 2002;20(3):719–26
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13(10):714–26
- Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–47
- Brufsky AM. Current Approaches and Emerging Directions in HER2-resistant Breast Cancer. Breast Cancer. 2014;61–7
- Rexer BN, Arteaga CL. Intrinsic and Acquired Resistance to HER2-Targeted Therapies in HER2 Gene-Amplified Breast Cancer: Mechanisms and Clinical Implications. Crit Rev Oncog. 2012;17(1):1–16.
- Creighton CJ. The molecular profile of luminal B breast cancer. Biol Targets Ther. 2012;6:289–97
- Wilkes EH, Casado P, Cutillas PR. Approaches for measuring signalling plasticity in the context of resistance to targeted cancer therapies. Biochem Soc Trans. 2014;42(4):791–7
- Cutillas PR. The role of proteomics in the development of personalised cancer medicine. 2013;18(2):47–50
- Targeted Cancer Therapies Fact Sheet – National Cancer Institute. [cited 2015 May 7]. Available from: http://www.cancer.gov/cancertopics/treatment/types/targeted-therapies/targeted-therapies-fact-sheet
- Chen K, Huang Y, Chen J. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin. 2013;34(6):732–40
- Britton D, Zen Y, Quaglia A, Selzer S, Mitra V, Loessner C, et al. Quantification of pancreatic cancer proteome and phosphorylome: Indicates molecular events likely contributing to cancer and activity of drug targets. PLoS One. 2014;9(3)
- Zen Y, Britton D, Mitra V, Brand A, Jung S, Loessner C, et al. Protein expression profiles of chemo-resistant mixed phenotype liver tumors using laser microdissection and LC-MS/MS proteomics. EuPA Open Proteomics. 2013;1:38–47
- Liu NQ, Braakman RBH, Stingl C, Luider TM, Martens JWM, Foekens J, et al. Proteomics pipeline for biomarker discovery of laser capture microdissected breast cancer tissue. J Mammary Gland Biol Neoplasia. 2012;17(2):155–64
- Casado P, Rodriguez-Prados J-C, Cosulich SC, Guichard S, Vanhaesebroeck B, Joel S, et al. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci Signal. 2013;6(268):rs6–rs6
- Casado P, Alcolea MP, Iorio F, Rodriguez-Prados J-C, Vanhaesebroeck B, Saez-Rodriguez J, et al. Phosphoproteomics data classify hematological cancer cell lines according to tumor type and sensitivity to kinase inhibitors. Genome Biol. 2013;14(4):R37
- Wilkes EH, Terfve C, Gribben JG, Saez-Rodriguez J, Cutillas PR. Empirical inference of circuitry and plasticity in a kinase signaling network. Pnas. 2015
Related topics
Analytical Techniques, Bioinformatics, Imaging, Kinases, Mass Spectrometry, Oncology, Proteomics, Therapeutics