Complex biology for the precision medicine era: re-thinking the drug discovery process
Posted: 4 September 2018 | Elad Katz | No comments yet
Complex biology is a discipline acknowledging that performing biological experiments in vitro should take account of the complexity of the biological context.1 While this may be a noble aim, it has proven difficult to incorporate these elements into the drug discovery process, especially at the high-throughput screening (HTS) stage.2
This is partly due to the difficulty of defining the boundaries of how complex biological systems need be to reflect the disease being targeted by drug discovery. Elements can stretch from the introduction of extracellular matrices (what is sometimes called 3D cell culture) and relevant media conditions (for example, specific cytokines instead of serum), all the way to co-culture with other cell types. However, beyond that lies a bigger problem in the context of HTS: which cells to use in the first place? The cells used in drug discovery have been diverse in range. Nonetheless, a large proportion of screens use several main workhorses, which have two main advantages: expandability and being easy to manipulate. These immortalised lines include CHO, HEK293, U2OS and HeLa cells. Despite their dependability, in expression of reporter genes, for example, these cell types are essentially irrelevant to most screens. Genetic manipulation of cell lines using CRISPR-based editing has undoubtedly expanded the range of cell lines available for HTS. However, certain cell types, such as lymphocytes, are still proving hard to manipulate.
To maintain the physiological relevance of the cells used, there is only one other resource to explore: the patients themselves. Patient-derived materials are diverse in nature and have traditionally been used as source material to generate cell lines by immortalisation. In recent years, methods of extracting cells have been improved by the introduction of commercial systems to isolate cells from tissues or tumours. Cell preservation methods have also improved, both by cryogenic storage and by passaging in mice (PDX models). However, these resources are relatively limited if immortalisation is avoided and cell numbers may be restricting any screen in size or inability to be used at hit-to-lead stages. This conundrum has led to two approaches using stem cell sources. One option is to use patient materials to generate inducible pluripotent stem cells (iPSCs) to reflect the variability of human disease.3 These cells need to be differentiated fully to the correct cell type – a process which typically takes weeks, if not months, and is not necessarily easy to scale up for HTS.
A more immediate solution is to derive organoids directly from the desired tissue. This effort has started in gut modelling, where the stem cell niche is well defined and has expanded to variable levels of success in other epithelial tissues. While direct pharmaceutical companies’ involvement has been limited thus far, several academic centres dedicated to organoids have now been established.4 Organoid-based (and probably iPSC-based) screening is still someway off being a plausible large-scale screening platform, not only due to limited cell numbers, but also because the read-outs are not entirely established. It is already fairly clear that natural variability will impact on important HTS factors, such as signal to noise ratio, as well as making any technical assay validation tricky.
The value in precision medicine-based screening
The growing appetite for precision medicine has not quite reached most screening laboratories, especially those outside academia. Nonetheless, the understanding that diseases are not homogenous is gradually seeping through and will, in time, affect every screening campaign. Whilst it is unlikely that there would ever be a primary screen devoted to each patient, the growing stratification of diseases into defined subtypes presents an opportunity to define parameters for screening that are both physiologically relevant and plausible on the large scale needed for screening. At the most basic level, there is awareness: what cell line is picked to screen against? Is it the best genetic match to the disease targeted? At a higher level, some genetic adaptation can be made to reflect a key factor in the disease. Another aspect, especially when working with iPSCs as source materials, is that the right developmental stage of tissue is reflected. Importantly, most iPSC-derived cells are foetal in characteristics, not adult.
One element commonly ignored during early drug discovery is the value that lies in obtaining biomarker data as part of the drug discovery process. The principle of precision medicine is applying the right drug to the right patients. If the prospective screen not only utilises relevant cells, but also relevant read-out (eg, secretion of relevant cytokines), this can be exploited further. This is especially true during lead optimisation when several read-outs can be tested in parallel on the basis of what was already used in the primary screen. Any complex biology-based screen in this context should provide viable biomarkers to carry over in the latter pre-clinical (and clinical) stages.
Case study: complex biology in screening for AML
To demonstrate the principles outlined above, I chose the emerging knowledge around acute myeloid leukaemia (AML). AML is a very heterogeneous blood cancer composed of at least 14 molecular sub-types, with ~58% of patients having an unfavourable outcome to current therapy.5 To screen for new targets within this space, a precision medicine approach is an absolute necessity. A pre-requisite for any meaningful screen necessitates a known association between the desired target and one or more AML subtypes, in the shape of expression, or preferably activity. This will define the cells to be used in any screening. Those can be derived directly from patients for a small-scale campaign or generated from an existing line, such as HL-60, by genetic manipulation to reflect the correct underlying mutation/s.6 Despite being small, this type of campaign can generate a multitude of meaningful read-outs including effects on proliferation, cell death and/or re-differentiation. While the introduction of stroma in screening will rightly be frowned on, there is now great in-depth knowledge of other factors that can make such screening more physiologically relevant to clinical AML, such as inclusion of key stroma-secreted cytokines such as IL-3, G-CFS.7
One option more aligned with current paradigms of small molecule HTS is to perform the primary screen as a standard enzymatic screen. The complex biology screen is then used as validation for enzymatic hits. This way it is broad enough to accommodate a range of possible chemistries, but also keeps tabs on the physiological context at a sufficiently early stage in the discovery process (Figure 1). Beyond hit-to-lead assessment, progress in the AML field suggests that lead optimisation can be performed in even more complex systems including stroma and extracellular matrices, provided read-outs are kept based on the cell biology used. Cytokine secretion is particularly attractive as it could essentially be read regardless of the cellular system. A great example of a complex biology platform in AML is a scaffold-based niche system described first in vitro and now replicated in vivo.8 This represents a new direction in which complex biology is introduced with greater accuracy during the drug discovery process to make sure the disease is well represented, thereby reducing the risk of failure in later pre-clinical, or indeed clinical, stages. An important advantage to this approach is the emergence of putative pharmacodynamic biomarkers as an integral part of the drug discovery process.
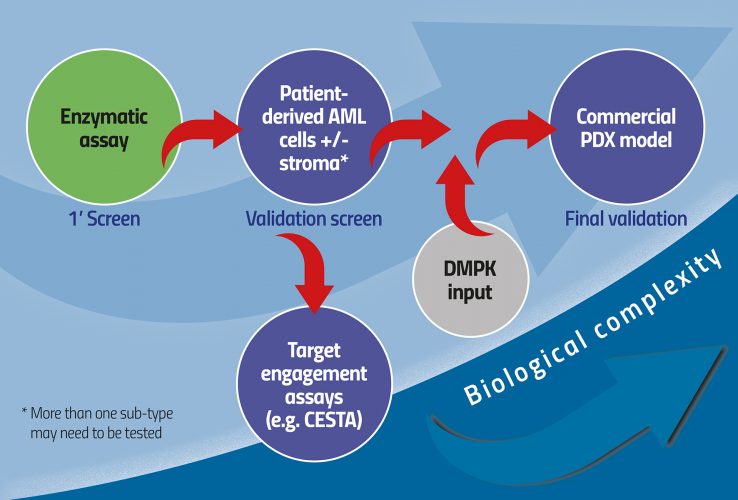
Figure 1: Increasing biological complexity from HTS to physiologically relevant models of AML
Conclusion
Here, I argue that it is possible to re-think the process of applying physiologically relevant inputs at the high-throughput setting by bridging target-based and phenotypic screening techniques. Specifically, by considering how specific disease cues can be integrated into cell-based high-throughput screens, their predictive value will be substantially increased. Furthermore, conversion of the screening process to a multi-read-out process in which medicinal chemists and protein engineers are considering more than one output to decide the value of a ‘hit’ will increase the value for money context in which the size of a prospective screen is decided. Taken together, incorporation of complex biology parameters into primary screens will increase their ultimate value and marry them more closely to questions of a mode of action and target validation than in the current common set-up.
References
- Chen CS. 3D Biomimetic Cultures: The Next Platform for Cell Biology. Trends Cell Biol. 2016;26(11):798-800.
- Horvath P, et al. Screening out irrelevant cell-based models of disease. Nat Rev Drug Discov. 2016;15(11):751-769.
- Kilpinen H, et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature. 2017;546(7658):370-375.
- Takebe T, et al. Organoid Center Strategies for Accelerating Clinical Translation. Cell Stem Cell. 2018;22(6):806-809.
- Papaemmanuil E, et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N Engl J Med. 2016;374(23):2209-2221.
- Bennett TA, et al. Pharmacological profiles of acute myeloid leukemia treatments in patient samples by automated flow cytometry: a bridge to individualized medicine. Clin Lymphoma Myeloma Leuk. 2014;14(4):305-18.
- Lam SS, et al. Treatment of acute myeloid leukemia in the next decade – Towards real-time functional testing and personalized medicine. Blood Rev. 2017;31(6):418-425.
- Abarrategi A, et al. Versatile humanized niche model enables study of normal and malignant human hematopoiesis. J Clin Invest. 2017;127:543-548.
Biography
DR ELAD KATZ is the inventor of Cancer-in-a-Box, a revolutionary cell biology device that can inform clinical treatment decisions for individual cancer patients. Since gaining his PhD from the University of Glasgow, Dr Katz has been working mainly on complex biology projects, in cancer and beyond. His team at the University of Edinburgh was first to show that tumour extracts can be preserved directly from human breast cancer to study drug responses ex vivo.
The rest of this content is restricted - login or subscribe free to access
 Thank you for visiting our website. To access this content in full you'll need to login. It's completely free to subscribe, and in less than a minute you can continue reading. If you've already subscribed, great - just login.
Thank you for visiting our website. To access this content in full you'll need to login. It's completely free to subscribe, and in less than a minute you can continue reading. If you've already subscribed, great - just login.
Why subscribe? Join our growing community of thousands of industry professionals and gain access to:
- quarterly issues in print and/or digital format
- case studies, whitepapers, webinars and industry-leading content
- breaking news and features
- our extensive online archive of thousands of articles and years of past issues
- ...And it's all free!
Click here to Subscribe today Login here
Related topics
Analysis, Assays, Cell Cultures, Cell-based assays, CRISPR
Related organisations
Cancer-in-a-Box