The implication of a dysfunctional proteasome regulator in neurodegenerative disease
Posted: 13 December 2019 | Hannah Balfour (Drug Target Review) | No comments yet
Disrupted protein homeostasis from PI31 inactivation may initiate the process of neurodegeneration seen in Alzheimer’s, ALS and other similar diseases, says new research.

Scientists at Rockefeller University in the US have established that knocking out a proteasome regulator, PI31, causes symptoms and morphological defects that closely model those seen in ALS patients. Drug Target Review’s Hannah Balfour explored the findings of the study and what it may mean for the future of neurodegenerative disease therapy research.
Scientists have long-ago established that neurodegenerative diseases such as Alzheimer’s Disease (AD) and Amyotrophic Lateral Sclerosis (ALS) are multi-factorial. Heavily studied and targeted in the hunt for a cure or prophylactic therapy for AD has been amyloid-β (Aβ), which constitutes part of the plaque pathology characteristic of the disease. But, why is there such a build-up of this protein to begin with?
The ubiquitin-proteasome system (UPS)
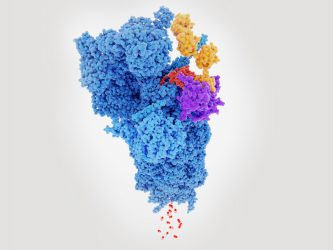
Proteasome degrading a protein (red) tagged with polyubiquitin (yellow).
Previous studies have established that UPS is responsible for the degradation of soluble intracellular proteins, vital to maintain protein homeostasis within many cell types, including neurones.
Biomarkers aren’t just supporting drug discovery – they’re driving it
FREE market report
From smarter trials to faster insights, this report unpacks the science, strategy and real-world impact behind the next generation of precision therapies.
What you’ll unlock:
- How biomarkers are guiding dose selection and early efficacy decisions in complex trials
- Why multi-omics, liquid biopsy and digital tools are redefining the discovery process
- What makes lab data regulatory-ready and why alignment matters from day one
Explore how biomarkers are shaping early drug development
Access the full report – it’s free!
Protein degradation by UPS has two stages; labelling, where a protein is tagged for destruction with a chain of ubiquitin molecules and then cleavage, where polyubiquitinated (poly-Ub) substrates are broken down into smaller peptides by the 26S proteasome. In neurodegenerative diseases, the function of UPS appears disrupted. This has been attributed to the ability of aggregates to ‘jam’ the proteasome complex. However, aggregate-mediated proteasome inhibition does not account for the failure to breakdown poly-Ub peptides prior to their aggregation.
Proteasome activity is highly regulated due to the high levels of damage that aberrant activity could cause. One regulator, the proteasome inhibitor of 31kD (PI31), is structurally conserved across species from Drosophila and yeast into mammals. However, there is conflicting evidence relating to its activity; initially discovered and named for having an ability to inhibit hydrolysis of small peptides by 20S proteasomes in vivo, in Drosophila, yeast and plants, it has since been seen to promote protein breakdown.
PI31 interacts with Tankyrase and the resulting modification promotes assembly of the 26S proteasome. It can also bind to a substrate recognition component of Skp, Cullin, F-box complex (SCF), a multi-protein E3 ubiquitin ligase complex that catalyses the ubiquitination of proteins destined for 26S proteasomal degradation. If the component, called Fbxo7 (PARK15 in humans), is absent then PI31 is cleaved and rendered inactive, reducing proteasome activity and causing neuronal degeneration.
Interestingly, mutations in Fxbo7/PARK15 are similarly associated with neurodegenerative diseases. Mutations in PARK15 have been identified in a severe form of early-onset Parkinson’s disease and mutations in the PI31 gene itself have been linked to Alzheimer’s disease.
Establishing the role of PI31
The researchers in the present study worked with PI31 and identified that, aside from its role in proteasome assembly, it is also an adapter for neuronal proteasome transport. The team used global and conditional knockout mice, of their own creation, to ascertain the role of PI31 in both spinal motor neurons (MNs) and cerebellar Purkinje cells (PCs). MNs and PCs were chosen for their association with numerous neurodegenerative diseases, including ALS, ataxias, autism and cerebellar essential tremor.
This is the first study to find a mechanism by which the proteasomes are moved to nerve endings to do their job”
In these knockout mice, the researchers established that PI31 is in fact a stimulant of protein breakdown. Furthermore, they concluded that it is essential for the maintenance of synaptic structure in long neurons because the first morphological defects in the conditional knockout model appeared in the axons and synapses. These irregularities were accompanied with a build-up of poly-Ub proteins in their distal synapses.
With age, the defects and symptoms became progressively worse. In the final stages of the disease, the axons and dendrites degenerated and neurons were lost. The symptoms in the mice included motoric problems which closely modelled those seen in patients with ALS.
“Notably, these structural changes became progressively more severe with age,” said Hermann Steller, head of the Strang Laboratory of Apoptosis and Cancer Biology at The Rockefeller University, and a Fellow of the American Association for the Advancement of Science. “Inactivation of PI31 in these neurons was reminiscent of the severe behavioural and anatomical defects we see in some human neurogenerative disease.”
The team also revealed that an inactivation of PI31 lead to a shortage of proteasomes at the synapses, with a resultant accumulation of poly-Ub proteins and aggregates. They concluded that this was due to the role of PI31 as an adapter protein, able to link proteasomes to cellular machinery for fast axonal transport. PI31 binds to dynein light chain proteins and proteasomes, acting as a coupling between the two.
Observing that inactivation of PI31 impaired proteasome motility, the researchers concluded this prevents proteasomes reaching the synapses of axons, thereby disrupting protein homeostasis in the distal reaches of neurons.
Steller highlighted: “This is the first study to find a mechanism by which the proteasomes are moved to nerve endings to do their job. When this mechanism gets disrupted, there are severe consequences for the function and long-term survival of nerve cells.”
The scientists concluded that since proper neuronal signalling requires effective synapse function including plasticity, which demands protein components be changeable, the neurons may be particularly sensitive to any disruption in protein homeostasis. This is only exacerbated in long neurons, where proteasome allocation and transport are more important. Therefore, the neuroprotective role of PI31 should not be overlooked.
Moving forwards
Steller and his team are now working to determine if PI31, or molecules that affect its activity, are viable drug targets. Given that PI31 appears to be involved in the early stages of nerve cell degeneration, drugs enhancing the activity of this protein might have the potential to halt neurodegeneration before it progresses to the stage of aggregation and neuronal loss.
…the researchers established that PI31 is in fact a stimulant of protein breakdown”
As for what causes brains to deteriorate, Steller believes the formation of aggregates is likely not the direct disease mechanism, but rather a symptom of bigger problems. “Our work suggests that it really starts with a local defect in proteasomes, resulting in the failure to degrade proteins that are critical for nerve function,” he says. “These undigested proteins subsequently form aggregates and activate additional damage control pathways. But eventually, these clearance systems are overwhelmed, which causes a slow but steady progression to a detectable disease.”
Steller and his colleagues are now investigating ways to stimulate the transportation pathway to get proteasomes out to the distal branches – work they believe could have a broad impact on the treatment of neurodegenerative diseases.
The findings were published in PNAS.
Related topics
Drug Targets, Neurons, Neuroprotection, Neurosciences, Targets, Therapeutics, Ubiquitin-proteasome system
Related conditions
Alzheimer’s disease, Amyotrophic Lateral Sclerosis (ALS), Neurodegenerative diseases
Related organisations
American Association for the Advancement of Science, Strang Laboratory of Apoptosis and Cancer Biology, The Rockefeller University
Related people
Hermann Steller