Exploiting functional selectivity of GPCRs in drug development
Posted: 21 September 2015 | Niklas Larsson (AstraZeneca), Torben Østerlund (AstraZeneca)
The large family of pharmaceutical targets represented by G-protein coupled receptors (GPCRs) are usually involved in a number of intracellular effector pathways, leading to many cellular outcomes. In recent years it has been uncovered that different ligands of a given GPCR may engage the pathways selectively, in that ligands that are equally potent in one pathway may have very different potencies in another.
The ligands are then said to be biased toward a pathway and induce functional selectivity at the receptor. This phenomenon can be utilised to develop selective ligand drugs with improved pharmacological properties or reduced adverse effects. We discuss examples of therapeutic advantages that such biased ligands may provide. We also describe how the desire to identify biased ligands affects the design of lead generation strategies and screening cascades.
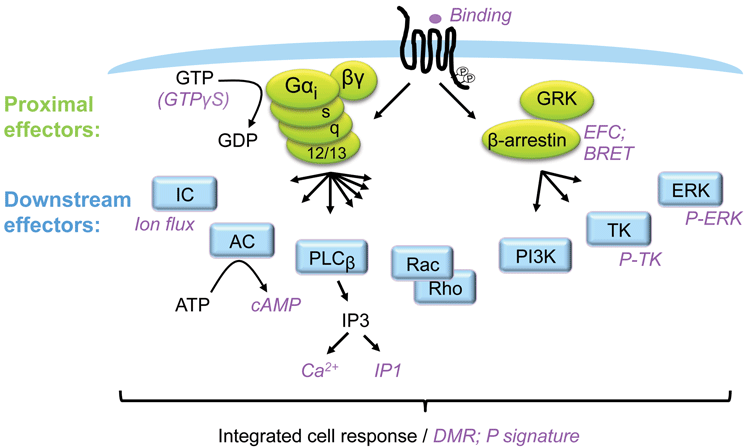
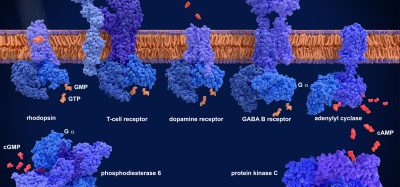
Figure 1: GPCR signalling and assays
GPCRs are the most explored family of pharmaceutical targets and because they usually couple to several intracellular signalling pathways, they control a variety of cellular functions. Frequently, the different pathways support overlapping biology. However, depending on cellular context, each pathway may serve a distinct function. GPCRs couple to G-proteins and β-arrestins as proximal effectors, and these proteins subsequently signal via a plethora of downstream effectors. It is possible to measure the engagement of the different pathways by measuring intracellular molecules accumulating or fading, with some of the common molecules measured downstream of GPCRs being cAMP, Ca2+, IP1 and pERK. Such assays will typically reveal the potency and efficacy of a ligand based on one (or a few converging) pathways.
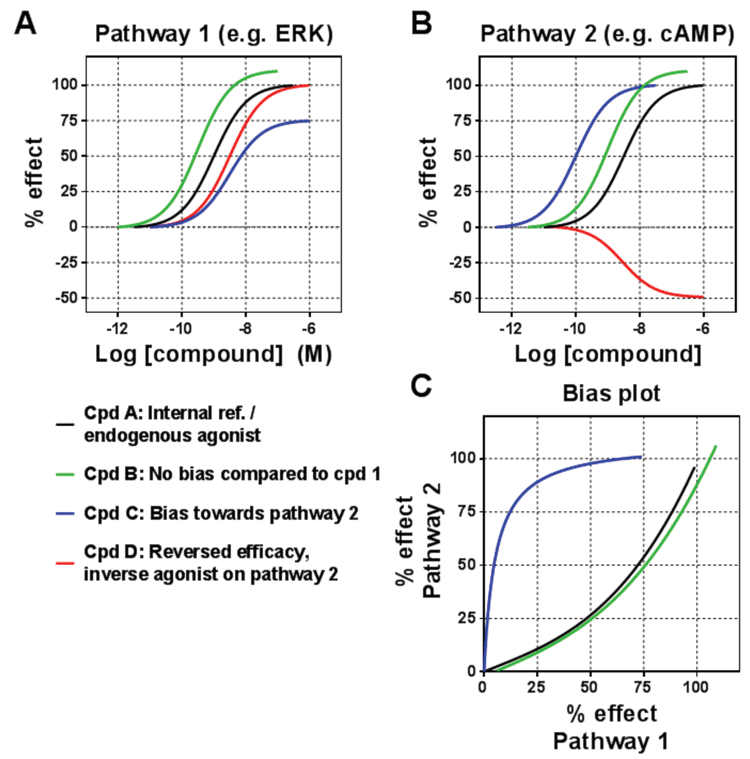
Figure 2: Measuring biased signalling
The traditional view was that receptor ligands would engage the different pathways similarly, with the same rank order of potency and efficacy irrespective of the pathway. However, this has frequently been challenged over the past two decades, with the phenomenon of functional selectivity (or ligand bias) becoming the terminology used to describe the way in which some ligands will engage the pathways in a different manner to other ligands. In some cases the engagement of pathways will have a different rank order of the ligands. This was first described in 1993 for the PACAP receptor, and then followed up by numerous examples.
Yet despite this building body of evidence, the phenomenon has only become widely accepted in more recent years, and it is now acknowledged that ligands may even selectively engage one pathway while not engaging another. This bias then translates into different functional consequences for the cells (and ultimately for the whole body) when ligands acting on the same receptor are compared with each other. In terms of the receptor-ligand interaction, biased ligands are selectively stabilising a subset of conformations in the conformational space representing the whole signalling repertoire of a receptor (Figure 3; page 00). Recent biophysical studies on the β2-adrenoceptor and V2 vasopressin receptor suggest that G-protein-biased and β-arrestin-biased ligands have distinct conformational effects supporting such a model4,5.
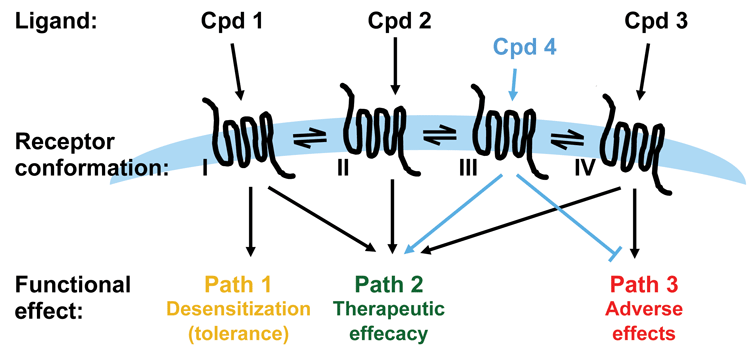
Figure 3: Applying functional selectivity in drug discovery
Considerations for drug discovery
In the realm of developing drugs that act on GPCRs, functional selectivity becomes an interesting concept. In the case where the pathway(s) determining the biological function of interest and pathway(s) determining unwanted or adverse effects are known, the possibility of developing ligands that select for the desired biological effect while deselecting the unwanted effects is intriguing. However, understanding the physiological role a specific pathway plays is also a big challenge for utilising functional selectivity in drug discovery. The ligands of a GPCR may not only differ in terms of the pathway selectivity as determined by cellular in vitro assays: they may in fact differ on a number of other parameters that affect the final outcome in vivo.
Each compound will inherently have specific drug metabolism and pharmacokinetic properties. These will contribute to the overall efficacy of, and adverse effects caused by, the compound and may explain, or contribute to, different pharmacodynamic effects of compounds quite separately from any functional selectivity at the receptor. This is important to consider when designing studies and several biased ligands may have to be tested in order to make a good case for functional selectivity. Thus, it is challenging to determine if a difference in biological outcome is due to the functional selectivity or due to, for instance, in vivo half-life, protein binding, exposure, bodily distribution, compartmentalisation or other phenomena in the animal models or patients in the clinic. It is also important to keep in mind that receptor and G-protein expression in the studied cellular system may contribute to artificial functional selectivity.
As discussed, it is critical to have a thorough understanding of the signalling pathways downstream of a receptor and their physiological and pathological effects before starting up a drug discovery program aiming to identify biased ligands. Whilst biased ligands present a great opportunity for drug discovery, a key challenge is building sufficient understanding of signalling pathways downstream of a receptor, and their crosstalk and impact on biology, in order to know which pathway in which cell/tissue to hit to achieve the desired therapeutic effect. Such knowledge generally calls for significant resources and time and will most likely require the identification of a number of ligands specific for the receptor being investigated. Downstream pathways such as those exemplified in Figure 1 (page 00) may have to be tested using selective inhibitors or expression knock-down technologies and availability of such is key to success. It is a clear advantage if cell and animal models for pathway selective biology are available. For example, β-arrestin knockout mice have been employed to build understanding of pathway-specific effects for a number of receptors, including the µ opioid receptor (MOR) discussed below6.
So-called β-blockers acting on β-adrenoceptors is an area where ligand-biased signalling has been extensively addressed7. An example of pathway selectivity can be illustrated by the compounds propranolol and ICI118,551. While these compounds are inverse agonists on the β2AR for cAMP production (similar to the majority of β-blockers), the latter act as partial agonists for ERK1/2 activation8,9. Such β-blockers with distinct pathway preferences have been compared in clinical trials. In a trial with chronic heart failure patients, carvedilol (β-blocker with partial ERK1/2 agonist activity) reduced mortality to a greater extent than metoprolol tartrate (β-blocker without ERK1/2 activity)10. However, it remains to be demonstrated if the difference in therapeutic effect was due to the ability of carvedilol to activate ERK1/2 or whether it was down to other factors. Interestingly, at the time carvedilol was designed, the phenomena of GPCR biased ligand or functional selectivity had not been described. It was only later that these pharmacological properties of carvedilol were discovered. In the light of current understanding, this example illustrates the challenge of establishing which pathways are relevant for clinical efficacy and adverse effects, and underscores the importance of addressing biased signalling when designing drug discovery strategies today. It also presents a potential opportunity to improve current GPCR-based therapies.
Screening approaches
Over the past decade, approaches have been developed to systematically identify and develop biased GPCR ligands1. Traditionally, single readout assays have been applied for hit finding/screening. A typical agonist cascade would contain a selected key pathway readout (e.g., cAMP) and a binding assay. To extend this approach for the identification of biased ligands, hits can subsequently be tested for activity in other pathway assays. Identified biased ligands then form the basis for new chemical design in order to explore the chemical space. However, such screening approaches are not able to capture the full repertoire of possible signalling responses and there is a risk that relevant compounds not potent on the primary pathway will be ignored. To minimise such a risk, screening cascades with multiple parallel assays measuring different end points have been employed. Although this gives a better chance of finding novel chemical starting points, the obvious drawback is the increased effort, time, cost and data complexity a broad parallel approach generates.
These drawbacks can to some extent be mitigated by measuring multiple signalling events (multiplexing) in a single assay. One helpful development is that of biosensors based on bioluminescence resonance energy transfer (BRET); these can be used for real-time monitoring of signalling events such as receptor-G protein interactions, production of second messengers and protein phosphorylation11. The list of such biosensors is continuously growing and some of these may be used for multiplexing applications.
However, in many cases one has to apply different technologies for different pathways. Such pathway-specific technologies suffer from their own artifacts and introduce a systematic bias when signalling through different pathways are compared. As an alternative to the use of multiple pathway-specific assays for primary screening, high throughput label-free technologies have been developed, which can give a holistic view of the cellular signaling events after receptor activation. The available label-free instruments either use impedance- or optical-based biosensors to detect changes in cell behaviour12,13. These technologies capture reorganisation of cellular components such as adhesion, cytoskeletal dynamics and overall changes in cell morphology. These events are often referred to as dynamic mass redistribution (DMR) and are a representation of the integrated cell response. It has been shown that optical biosensors are able to capture signalling via all major classes of G-proteins, have sufficient sensitivity to study endogenously expressed GPCRs in primary cells14 and are amenable to HTS15.
Systematic development of biased ligands resulting in compounds reaching the clinic
Compounds where systematic optimisation of biased signalling properties has been applied are now in clinical trials. One example is the angiotensin II type 1 receptor (AT1R) that signals through both Gαq- and β-arrestin-dependent pathways and is a well characterised system of functional selectivity16. Key data underlining the value of a biased ligand approach for this target include investigations of [Sar1,Ile4,Ile8]-angiotensin (SII), which activates β-arrestin2 signalling while not engaging G-protein signalling. It has been shown that SII stimulates contractility of cardiomyocytes via AT1R in a β-arrestin2-dependent manner17. Such observations together with data from additional cell and animal models early on led Lefkowitz and colleagues to hypothesise that it should be possible to develop a novel therapy for cardiovascular disease by optimising SII-like compounds that maintain the positive renal and vascular effects of an angiotensin receptor (Gαq pathway) blocker. It was also supposed that additional beneficial effect could come from enhancing myocontractility and cell survival, provided by activation of the β-arrestin/ERK pathway18,19. This work has culminated in the development of TRV027 (Trevena, Inc.), currently in Phase 2b clinical trials for the treatment of acute heart failure.
Another example is provided by the µ opioid receptor (MOR). µ opioid agonists such as morphine and analogues are highly efficacious analgesics acting through MOR. However, MOR agonists generally have severe adverse effects including nausea, respiratory depression, sedation and abuse. Unfortunately, these side effects are also mediated via MOR20. Different approaches such as peripheral restriction and partial agonism have been employed to develop MOR agonists with reduced adverse effects21,22. Although successful for specific applications, on the receptor level these approaches do not distinguish analgesia from undesired effects. Experiments in β-arrestin2 knockout mice have helped to dissect pathway components important for analgesia and adverse effects. Simplified, data suggest that MOR stimulation of Gαi results in analgesia whereas mainly β-arrestin2 recruitment and associated downstream events are responsible for desensitisation, tolerance and adverse effects6,23,24. Lead discovery and optimisation efforts looking for biased MOR agonists have led to generation of Trevena’s candidate TRV130, which is a small molecule Gαi biased agonist with very low β-arrestin recruitment efficacy25. In rodents, TRV130 is analgesic while causing less gastrointestinal dysfunction and respiratory suppression than morphine at equianalgesic doses26. This compound is now in clinical trials and being tested head-to-head against morphine, which will be very interesting to follow.
Concluding and future perspectives
Biased GPCR ligands with functional selectivity represent exciting opportunities for drug discovery, opportunities that have only been exploited to a limited degree. With respect to hit finding and in vitro pharmacology, strategies and technologies to identify and characterise biased ligands are well developed. In fact, it appears to be highly feasible to find biased ligands for many GPCRs. The most prominent challenge working with biased ligands is rather to select or develop the most appropriate biological systems to study the effects of functional selectivity, since the effects are context specific. Since a ligand may show bias in one cell system but not in another, it is key to use a system, preferentially a primary cell or in vivo system, where the action can specifically be ascribed to the receptor and pathway of interest. Without appropriate in vivo models, it is challenging to verify that a specific biological outcome is due to pathway bias or to other parameters differing between the compounds. So far, mice models such as β-arrestin knockouts have been very valuable for pathway validation. Being in the middle of the CRISPR/Cas9 craze we expect that development of additional clever and more complex cellular and animal models facilitating pathway deconvolution will continuously be generated.
In this mini-review, we have mainly discussed G-protein versus β-arrestin bias. However, examples are emerging of ligand bias with respect to activation of specific subsets of G-proteins as well27.Together with new attractive ways to investigate functional selectivity such as measuring the phosphorylation ‘barcode’ on a receptor28,29, or phosphoproteomics to investigate compound-induced changes in signalling cascades in relevant tissues after compound treatment30, this further extends the scope of functional selectivity.
To summarise, the idea of selectively modulating the activity of a signalling pathway relevant for a disease or treatment whilst reducing other receptor-mediated mechanisms important for adverse or irrelevant effects is very attractive. The ongoing clinical evaluations of biased agonists for different indications suggest that future drug development efforts may increase the focus on assessing functional selectivity as a way to develop safer and more efficacious medicines.
References
- Stallaert W, Christopoulos A, Bouvier M. Ligand functional selectivity and quantitative pharmacology at G protein-coupled receptors. Expert Opin Drug Discov. 2011 Aug;6(8):811-825
- Spengler D, Waeber C, Pantaloni C, Holsboer F, Bockaert J, Seeburg PH, et al. Differential signal transduction by five splice variants of the PACAP receptor. Nature. 1993 Sep 9;365(6442):170-175
- Whalen EJ, Rajagopal S, Lefkowitz RJ. Therapeutic potential of beta-arrestin- and G protein-biased agonists. Trends Mol Med. 2011 Mar;17(3):126-139
- Liu JJ, Horst R, Katritch V, Stevens RC, Wuthrich K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. 2012 Mar 2;335(6072):1106-1110
- Rahmeh R, Damian M, Cottet M, Orcel H, Mendre C, Durroux T, et al. Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc Natl Acad Sci USA. 2012 Apr 24;109(17):6733-6738
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999 Dec 24;286(5449):2495-2498
- Patel CB, Noor N, Rockman HA. Functional selectivity in adrenergic and angiotensin signaling systems. Mol Pharmacol. 2010 Dec;78(6):983-992
- Azzi M, Charest PG, Angers S, Rousseau G, Kohout T, Bouvier M, et al. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc Natl Acad Sci USA. 2003 Sep 30;100(20):11406-11411
- Baker JG, Hall IP, Hill SJ. Agonist and inverse agonist actions of beta-blockers at the human beta 2-adrenoceptor provide evidence for agonist-directed signaling. Mol Pharmacol. 2003 Dec;64(6):1357-1369
- Metra M, Cas LD, di Lenarda A, Poole-Wilson P. Beta-blockers in heart failure: are pharmacological differences clinically important? Heart Fail Rev. 2004 Apr;9(2):123-130
- Salahpour A, Espinoza S, Masri B, Lam V, Barak LS, Gainetdinov RR. BRET biosensors to study GPCR biology, pharmacology, and signal transduction. Front Endocrinol (Lausanne). 2012 Aug 29;3:105
- Scott CW, Peters MF. Label-free whole-cell assays: expanding the scope of GPCR screening. Drug Discov Today. 2010 Sep;15(17-18):704-716
- Larsson N, Sundström L, Ryberg E, Frostne L. Cell based label-free assays in GPCR drug discovery. 2013;18(4):13
- Schroder R, Janssen N, Schmidt J, Kebig A, Merten N, Hennen S, et al. Deconvolution of complex G protein-coupled receptor signaling in live cells using dynamic mass redistribution measurements. Nat Biotechnol. 2010 Sep;28(9):943-949
- Dodgson K, Gedge L, Murray DC, Coldwell M. A 100K well screen for a muscarinic receptor using the Epic label-free system–a reflection on the benefits of the label-free approach to screening seven-transmembrane receptors. J Recept Signal Transduct Res. 2009;29(3-4):163-172
- Aplin M, Christensen GL, Hansen JL. Pharmacologic perspectives of functional selectivity by the angiotensin II type 1 receptor. Trends Cardiovasc Med. 2008 Nov;18(8):305-312
- Rajagopal K, Whalen EJ, Violin JD, Stiber JA, Rosenberg PB, Premont RT, et al. Beta-arrestin2-mediated inotropic effects of the angiotensin II type 1A receptor in isolated cardiac myocytes. Proc Natl Acad Sci USA. 2006 Oct 31;103(44):16284-16289
- Rajagopal K, Lefkowitz RJ, Rockman HA. When 7 transmembrane receptors are not G protein-coupled receptors. J Clin Invest. 2005 Nov;115(11):2971-2974
- Violin JD, Lefkowitz RJ. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci. 2007 Aug;28(8):416-422
- Kieffer BL, Gaveriaux-Ruff C. Exploring the opioid system by gene knockout. Prog Neurobiol. 2002 Apr;66(5):285-306
- Taguchi A, Sharma N, Saleem RM, Sessler DI, Carpenter RL, Seyedsadr M, et al. Selective postoperative inhibition of gastrointestinal opioid receptors. N Engl J Med. 2001 Sep 27;345(13):935-940
- Lutfy K, Cowan A. Buprenorphine: a unique drug with complex pharmacology. Curr Neuropharmacol. 2004 Oct;2(4):395-402
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000 Dec 7;408(6813):720-723
- Raehal KM, Walker JK, Bohn LM. Morphine side effects in beta-arrestin 2 knockout mice. J Pharmacol Exp Ther. 2005 Sep;314(3):1195-1201
- Chen J, Kuei C, Sutton SW, Bonaventure P, Nepomuceno D, Eriste E, et al. Pharmacological characterization of relaxin-3/INSL7 receptors GPCR135 and GPCR142 from different mammalian species. J Pharmacol Exp Ther. 2005 Jan;312(1):83-95
- DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, et al. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013 Mar;344(3):708-717
- Busnelli M, Sauliere A, Manning M, Bouvier M, Gales C, Chini B. Functional selective oxytocin-derived agonists discriminate between individual G protein family subtypes. J Biol Chem. 2012 Feb 3;287(6):3617-3629
- Doll C, Konietzko J, Poll F, Koch T, Hollt V, Schulz S. Agonist-selective patterns of micro-opioid receptor phosphorylation revealed by phosphosite-specific antibodies. Br J Pharmacol. 2011 Sep;164(2):298-307
- Butcher AJ, Hudson BD, Shimpukade B, Alvarez-Curto E, Prihandoko R, Ulven T, et al. Concomitant action of structural elements and receptor phosphorylation determines arrestin-3 interaction with the free fatty acid receptor FFA4. J Biol Chem. 2014 Jun 27;289(26):18451-18465
- Hoffert JD, Pisitkun T, Saeed F, Song JH, Chou CL, Knepper MA. Dynamics of the G protein-coupled vasopressin V2 receptor signaling network revealed by quantitative phosphoproteomics. Mol Cell Proteomics. 2012 Feb;11(2):M111.014613
- Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2013 Mar;12(3):205-216
Biographies
Dr. Niklas Larsson obtained his PhD in 1999 in Cell and Molecular Biology at Umeå University, Sweden. Since 2000 he has been working with early drug discovery projects at AstraZeneca (Mölndal, Sweden) in different positions such as Project Leader, Team Leader and currently as Associate Principle Scientist within the Discovery Sciences function. His scientific focus is drug discovery and molecular pharmacology of GPCRs.
Dr. Torben Østerlund obtained his PhD in 1998 in Cell and Molecular Biology from the Medical Faculty at Lund University, Sweden. He has since worked with cellular signalling in both cancer and the diabetes/metabolism areas. Since 2008 he has worked in pharma/biotech with cell biology and signalling. In 2010 he joined Zealand Pharma working with molecular pharmacology of GPCRs including functional selectivity. He joined AstraZeneca in 2014 as Senior Scientist with a specialty in cell assays and molecular pharmacology.
Related topics
Assays, Cell-based assays, GPCRs
Related organisations
AstraZeneca
Related people
Niklas larsson, Torben Østerlund