

Immunogenicity: anticipating and avoiding issues for biopharmaceutical development
Posted: 21 September 2015 | Paul Chamberlain (NDA Advisory Board)
All biopharmaceutical products are associated with an intrinsic potential to induce immune responses in treated subjects. Regulatory agencies expect sponsors to evaluate and mitigate these risks during product development, applying a strategy that addresses product- and patient-related factors. Overall, understanding and controlling immunogenicity-related risks are attainable objectives, and approvability should not be compromised if these risks are suitably evaluated from the earliest stages of development.

Immunogenicity refers to the capacity of a drug product to induce an immune response in the recipient: this may be the intended effect, as in the case of vaccines, or unintended. Depending on the nature and magnitude of the response, unintended immunogenicity can impact negatively on overall clinical benefit and risk.
All biopharmaceutical products have an intrinsic immunogenic potential by virtue of molecular features that can stimulate cells of the adaptive and innate immune systems. However, this does not imply that immunogenicity will have a negative influence on treatment outcome – rather, the challenge for the regulator is to understand the scale of the immune response in relation to risk of adverse events and/or reduced efficacy for the population to be treated.
Accordingly, the critical question to address for developability and approvability is whether manifestations of an unintended immune response to the drug product are of a sufficient scale – taking into account the rate of occurrence and the severity of consequences – to affect the overall clinical benefit and risk balance. In the vast majority of cases, unintended immunogenicity does not represent a roadblock for successful clinical development and registration. Moreover, sponsors can effectively de-risk biopharmaceutical development by anticipating and mitigating immunogenicity-related risks.
Method of evaluation
At the lead candidate selection stage, in silico and in vitro analytical tools can be applied to assess the relative immunogenic potential in human subjects1, and a manufacturability analysis would include a thorough review of strategies that are being used to control product-, process- and primary-container-related risk factors.
Depending on the extent of molecular features shared with endogenous counterparts, clinical trial-enabling studies may be designed to evaluate potential clinical consequences of unintended immune responses within the context of hazard identification to mitigate risks for first administration to human subjects. Conception of an adaptive bioanalytical strategy that is aligned to detect product-specific risks is also part of this stage.
Correlation of bioanalytical signals of immunogenicity with relevant clinical endpoints during early clinical development instructs the decision to proceed into confirmatory clinical studies. The Sponsor must put careful thought into the clinical study design, selecting suitable bioanalytical methods and a sampling schedule for monitoring immune responses to the product, in order to address the regulator’s priorities. All too often, the extent of testing is insufficient to evaluate potential for pre-sensitised subjects to react to components of the drug product, or of treatment-emergent anti-drug antibodies to cross-react with endogenous factors. Definition of the dynamics of the immune response is crucial in identifying associations with clinically meaningful effects.
For registration, the Sponsor’s presentation of an integrated data summary enables regulatory reviewers to make a multi-disciplinary assessment of the impact of unintended immunogenicity on overall clinical benefit and risk in the target population(s). The data summery should be structured to facilitate interpretation of clinical observations relative to intrinsic immunogenic potential, systems biology, product quality- and patient-related factors. The conclusions of the integrated summary also serve to justify the proposed provisions in the Risk Management Plan to mitigate uncertainty in the post-authorisation setting.
Overall, understanding and controlling immunogenicity-related risks are attainable objectives, and approvability should not be compromised if these risks are suitably evaluated from the earliest stages of development.
Case example: a PEGylated enzyme replacement therapy
This case example concerns a modified version of agalsidase (recombinant human a-galactosidase), which is PEGylated to extend half-life in the systemic circulation without affecting uptake into target cells. The protein is expressed in a tobacco host cell, which introduces non-human glycans, and then conjugated to a bifunctional 10 kDa PEG (see Figure 1).
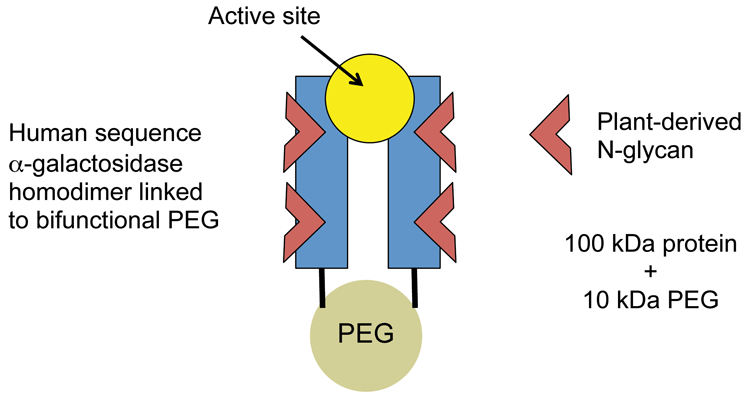
Figure 1: PEGylated agalsidase (plant-derived)
Enzyme replacement therapies are used to treat subjects with a deficiency in a naturally occurring enzyme arising from different types of genetic mutations (e.g., point, missense or deletion). The extent of deficiency of the native enzyme is often related to the extent of genetic polymorphism; patients with the most severe deficiencies tend to have lower immune tolerance to an exogenous enzyme replacement therapy, with the consequence that they mount an immune response to the therapeutic protein, which can compromise treatment outcome.
Indeed, this is the case for FabrazymeÒ (agalsidase) itself, which is approved for the treatment of Fabry disease. In pre-authorisation clinical studies for Fabrazyme, the majority of patients developed anti-drug antibodies (ADA) of the IgG class, although no antigen-specific IgE was detected2; some 50% of subjects developed mild-to-moderate adverse events on the day of infusion, but were able to continue treatment using a slower infusion rate and/or pre-medication. Complement activation was detected in some patients, but there was no evidence of immune-complex mediated glomerulonephritis on renal biopsy.
Minimising impact of immunogenicity
To clinically evaluate immunogenicity-related risks with the PEGylated agalsidase product candidate, a risk-based approach3 should focus on evaluating the relationship between genotype, phenotype and the treatment-emergent immune response in individual subjects. The following data outputs would be relevant:
- Genotyping;
- Suitably-specific and sensitive bioanalytical methods to monitor the immune response and to measure levels of the endogenous enzyme (in plasma and in leukocytes);
- Biomarkers of clinical response; and
- Effective integration of bioanalytical signals with clinical endpoints and with genotype.
There are important confounding factors for the assessment, including the relatively low number of subjects for clinical evaluation as well as a high degree of genetic polymorphism. On the other hand, the importance of undesirable immunogenicity on overall clinical benefit-to-risk would be moderated by the severe morbidities associated with the disease.
Since the PEGylated agalsidase has a modified molecular structure compared with Fabrazyme, it would be relevant to demonstrate the lack of incremental risk associated with the modifications, most notably:
- Absence of allergenicity associated with plant-derived glycans; and
- No increased immunogenicity associated with longer half-life of PEGylated version, or with PEG-reactive antibodies.
Clearly, thorough analytical characterisation to define levels of plant-derived glycans, other product-related variants, and process-related impurities (including tobacco cell-derived protein co-purifying with agalsidase) in the clinical and commercial-grade drug product would represent an important aspect of the justification of product specifications. Whilst it would not be necessary to perform a comparative clinical study of the PEGylated versus non-PEGylated agalsidase, it might be instructive to perform a comparative immunogenicity study in a relevant animal model, to demonstrate an absence of a marked increase in immunogenicity of the PEGylated product.
Another issue to consider is that although the potential risks should be evaluated for each new product candidate, it is relevant to cross-refer to experience gained for other products that share common risk factors. For example, while production of therapeutic proteins in plants has been associated with a theoretical risk of immunogenicity/allergenicity due to recognition of non-human glycans, this risk was not substantiated by the experience gained with plant-derived virus-like particles4. Nevertheless, the design of IND-enabling and clinical studies should be adapted to address the theoretical risk, since the scale of risk could be influenced by a multitude of factors.
Evaluation during IND-enabling studies
Based on the identified immunogenicity-related risk factors for the PEGylated agalsidase product, it would be relevant to include a number of evaluative studies, as well as implementing appropriate product quality controls, prior to initiating clinical studies (see Table 1; page 00).
The Fabry mouse model5 could be used for direct comparison of the relative immunogenicity of PEGylated and non-PEGylated versions in the context of a phenotype lacking endogenous alpha-galactosidase expression; adapted versions6 are also able to recapitulate symptomology of Fabry disease in humans, enabling measures of relative immunogenicity to be correlated with pharmacokinetics (PK) and pharmacodynamics.
The results may be sufficiently informative to demonstrate an absence of incremental risk of immunogenicity of the PEGylated, plant-derived enzyme compared with the non-PEGylated version expressed in Chinese hamster ovary cells. Bioanalysis of treatment-induced ADA responses could exclude reactivity with pre-existing antibodies with cross-reactivity to un-related proteins expressed in tobacco cells (expressing common plant-derived glycans), as well as exclude induction of treatment-emergent antibodies reactive with plant-derived glycan.
Ex vivo basophil activation testing using cells isolated from sensitised human subjects in comparison with cells from normal human subjects provides a sensitive system for preclinical evaluation of allergenic risk. This test also has higher diagnostic specificity than measurement of total or antigen-specific IgE.
Early clinical development
Bioanalysis of pre- and post-treatment samples from Phase 1 and Phase 2 clinical studies would focus on the addressing the dynamics and specificity of product-reactive antibodies relative to tolerability. In particular, these studies could provide preliminary evidence of whether there was a real risk of allergic reactions, as well as revealing a relationship between the extent of deficiency of the endogenous enzyme and the immune response to the exogenous therapeutic protein – which could reflect a lower level of immune tolerance in more severely affected subjects, or an influence of disease morbidity on immune responsiveness.
Observations of potential allergic reactions should trigger follow-up investigations in affected subjects, including ex vivo basophil activation testing and measurement of antigen-specific IgE. Controls should include an unrelated protein expressed in tobacco cells, and bearing a similar qualitative composition of plant-derived N-glycans as well as agalsidase expressed in a mammalian cell system.
Table 1: Preclinical risk evaluation for PEGylated agalsidase (plant-derived)
|
Activity |
Evaluation |
|
Develop & qualify bioanalytical methods |
Endogenous agalsidase protein Exogenous agalisidase protein enzyme activity Anti-agalsidase antibodies (ADA) |
|
Comparative immunogenicity in vivo e.g. Fabry mouse
|
Compare immune response for PEG-agalsidase (plant) vs. agalisdase (CHO): ADA IgG titer Inhibition of target cell uptake by ADA Cross-reactivity of ADA with different antigens;
|
|
Repeat-dose Toxicokinetics |
Correlate indices of ADA IgG vs. PK vs. Complement activation / immune-complex deposition |
|
Allergenic potential ex-vivo |
Ex vivo antigen challenge of basophils from normal subjects vs. pre-sensitized to plant glycans |
|
Product quality control |
Base initial release specifications on levels of on non-human glycans detected in preclinical batches |
Comparison of PK parameters at the first and final doses would also enable estimation of the extent of ADA response on circulating drug levels. Arguably, a functional assay of neutralising capacity of ADAs in plasma would have equivocal relevance because the enzyme acts intracellularly in the acidic conditions of the lysosomal compartment. Instead, it may be more relevant to evaluate whether the presence of ADAs can impair cellular uptake of PEGylated agalsidase.
In a few cases, clinically relevant PEG-reactive antibodies have been induced by treatment with PEGylated therapeutic proteins (PEGylated uricase and PEGylated asparaginase) for which there was incomplete immune tolerance allied to co-morbidities that may have presented ‘danger’ signals that enhanced immune responsiveness to the PEG moiety7. Pre-existing antibodies induced by dietary or cosmetic exposure to products containing PEG can confound detection of treatment-emergent PEG-reactive antibodies. Accordingly, the ADA assay should be validated for specificity and sensitivity to detect antibodies reactive with the PEG moiety of PEGylated agalsidase.
Pivotal clinical studies
Since eligible subjects may have previously been treated with other agalsidase products, and this could impact on immunogenicity of a subsequently administered product containing agalsidase, it would be relevant to include both previously-treated and treatment-naïve subjects in a randomised controlled trial of PEGylated agalsidase versus best current standard of care (see Figure 2).
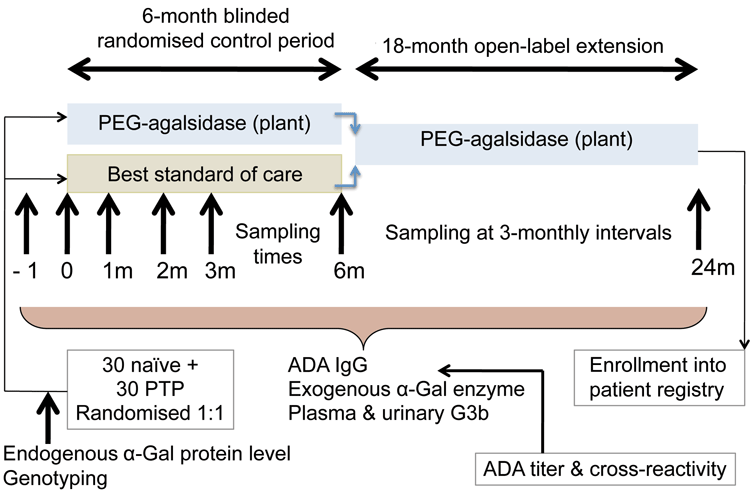
Figure 2: Immunogenicity risk evaluation for PEGylated agalsidase in Phase 3 study
Monitoring of immunogenicity during a 6-month treatment period would enable the dynamics of ADA formation to be related to baseline endogenous alpha-galactosidase, steady state drug trough concentration, PD (plasma and urinary G3b), disease status and the incidence and severity of immune-mediated adverse events.
All subjects could then be eligible for continuing treatment during an open-label extension period, during which immunogenicity monitoring would be performed to define impact on longer-term treatment outcomes. Ideally, the subjects completing treatment could then be included in a patient registry that monitored the response to continuing treatment.
Managing regulatory expectations
Regulators consider risk in relation to benefit: this means that the scale of regulatory concern about the undesirable immunogenicity of any given product candidate will always be dependent on the balance between the severity and probability of adverse effects versus the favourable outcomes.
Sponsors must provide sufficient evidence to enable the regulator to reach an informed decision on the overall benefit to risk proposition. Moreover, since there will be residual risk and uncertainty about the scale of the risk, it is important to understand the nature of the risk and how this risk might manifest in the population(s) to be treated – thereby, informing monitoring and risk mitigation measures. The regulatory principle is: “knowing risks means controlling risks”.
For a dossier supporting an application to perform a clinical study, it is helpful to provide the regulatory reviewers with a clear description of the risks that the sponsor has considered, taking into account intrinsic and extrinsic factors that are relevant for the product and the proposed target population. This information could be included as an Appendix to the IND or IMPD, and cross-referenced in the justification of overall benefit-to-risk. Summary information on the design and performance of bioanalytical methods is also valuable in demonstrating that the sponsor will be applying suitable methodology to evaluate the relevant risks in controlled clinical studies.
The MAA or BLA dossier could incorporate an ‘Integrated Summary of Immunogenicity’ (ISI) in section 5.3.5.3, whose format is designed to address a comprehensive multi-disciplinary review for marketing authorisation8. Ideally, this would present a complete storyline, starting from an introduction of the intrinsic immunogenic potential of the product, systems biology and extrinsic factors, and then followed by data correlations to support conclusions regarding clinical impact of detected immune responses. Individual patient data could be included in appendices, thereby providing the reviewers with a self-standing module. The ISI could also provide an explicit linkage to the proposed ongoing risk management provisions.
Certainly, starting out at the lead candidate stage with this format in mind can be extremely helpful in building a suitably thorough non-clinical and clinical evaluative strategy that is linked to product attributes and intended clinical use. The initial risk assessment document can be updated at each successive stage-gate according to the particular risks identified, and the data accumulated to address these. Transparency is provided to the product development team, who may then adapt planned activities according to regulatory need.
References
- Jawa V, Cousens LP, Awwad M, Wakshull E, Kropshofer H, De Groot AS. T-cell dependent immunogenicity of protein therapeutics: Preclinical assessment and mitigation. Clin Immunol. 2013; 149, 534-555
- European Public Assessment Report (EPAR) for Fabrazyme®, available at ema.europa.eu
- Buettel IC et al. Taking immunogenicity assessment of therapeutic proteins to the next level. Biologicals. 2011; 39 (2), 100-109
- Ward BJ et al. Human antibody response to N-glycans present on plant-made influenza virus-like particle (VLP) vaccines. Vaccine. 2014; 32, 6098-6106
- Garman RD, Munroe K, Richards SM. Methotrexate reduces antibody responses to recombinant human a-galactosidase A therapy in a mouse model of Fabry disease. Clin Exp Immunol. 2004; 137, 496-502
- Taguchi A et al. A symptomatic Fabry disease mouse model generated by inducing globotriaosylceramide synthesis. Biochem J. 2013; 456 (3), 373-383
- Verhoef JJ, Carpenter JF, Anchordoguy TJ, Schellekens H. Potential induction of anti-PEG antibodies and complement activation toward PEGylated therapeutics. Drug Discov Today. 2014; 19 (12), 1945-52
- Chamberlain P. Addressing immunogenicity-related risks in an integrated manner. Regulatory Affairs Pharma. Jan 2011; 10-15
Biography
Paul Chamberlain has been a Member of NDA Advisory Board since 2007. Previously, he was Senior Director of Biopharmaceutical Development Programs at MDA Pharma Services. He has prepared immunogenicity risk management strategies for diverse product types, including therapeutic proteins and peptides, advanced therapy medicinal products and medicinal product-device combinations. He specialises in the presentation of immunogenicity-related data to regulatory agencies to support applications for clinical trials and marketing authorisation.
