Next-generation transcriptomic analysis in cancer vascular research
Posted: 3 December 2015 | Joseph Wragg (University of Birmingham), Roy Bicknell (University of Birmingham) | No comments yet
Over the past decade significant advances have been made in the fields of genomic and transcriptomic profiling, inspired by the advent of next-generation sequencing (NGS). Yet despite the considerable promise of these new technologies, uptake has been slow. The focus of this review is the use of next-generation transcriptomic analysis in the field of cancer endothelial biology, highlighting its advantages and a few of the disadvantages compared with current-generation technologies.
NGS is an emerging technology for genomic, epigenomic and transcriptomic profiling, which can be performed more rapidly than traditional Sanger sequencing and in far greater depth than microarray technologies1. NGS technologies have been used in a wide variety of fields from sequencing of bacterial and viral genomes2,3, searching for patient-specific genetic variations4 and profiling of DNA-binding proteins by ChIP-Seq5, to characterising the transcriptomes of cells, tissues and organisms by RNA-Seq6. NGS covers a wide variety of technologies but primarily operates by sequencing DNA or RNA samples in a massively multiplexed manner, generating images, from which short sequence reads can be determined, aligned to a reference genome and converted to genomic data7.
Tumours are highly dependant on their vasculature to support their growth, being unable to expand beyond 2mm in size or metastasise in the absence of vessels to supply nutrients, remove waste products and propagate tumour cells around the body8,9. Considerable effort has been expended to investigate the mechanisms by which tumour vasculature develops, as well as examining its distinctiveness from the vessels of healthy tissues, with the aim of depriving the tumour of its blood supply. The vast majority of transcriptomic analysis involved in this work has been performed using microarray technologies, though SAGE and cDNA library analysis have also been used (reviewed in10). The advent of highthroughput next-generation analysis offers the opportunity for new discoveries to be made in this field, discoveries which are not possible with older technologies, and this will contribute to refinement and improvement of vessel-targeted anti-cancer therapies.
RNA-Seq to study antiangiogenic therapies
Cancer vessel-targeted therapies broadly divide into two categories; antiangiogenic therapies, which aim to block the processes by which cancers vascularise11 and vascular disrupting agents, which aim to destroy existing vessels in the tumour12. Antiangiogenics are the better-established therapeutic intervention, contributing several therapies to the clinic, including Sutent® (sunitinib), Nexavar® (sorafenib), cediranib (in development by AstraZeneca) and Avastin® (bevacizumab), which have contributed considerably to survival in several cancer types (reviewed in 10).
However, the efficacy of this approach has been curtailed by difficulties of acquired and innate resistance (reviewed in 10,13). Transcriptomic analysis techniques have been utilised in the search for the molecular mechanisms behind resistance and for markers of response to these therapies, to better tailor the use of these drugs to the most profitable indication (reviewed in 14).
The majority of this work has been performed using present-generation technologies. Next-generation technologies do offer some advantages, however. For example, Illumina RNA-Seq NGS was used to genetically profile the tumour from a patient exhibiting an unusually positive response to Sunitinib in a clinical trial of testicular cancer. This study identified the amplification of several candidate markers of response to Sunitinib including RET, EGFR and KRAS15. This study was part of a wider program at the MD Anderson Cancer Center in Texas, profiling abnormal responders to therapeutics using next-generation genomic technologies, with the aim of identifying novel markers of drug response or resistance16.
RNA-Seq differs from traditional transcriptomic analyses such as probe-based microarray systems in that rather than providing a relative expression level of a candidate gene via comparative fluorescence signal analysis, NGS provides an absolute expression level. This is undertaken by sequencing the prepared genetic material from the sample and determining the copy number of transcripts corresponding to candidate genes. In the example given, the use of NGS technologies allowed the identification of genes that were not just differentially expressed but also present in sufficient quantities to be clinically relevant for further investigation15. Thus RNA-Seq has overcome one of the weaknesses of microarray-based analysis, which identify a huge number of differentially expressed genes, most of which are present in such minuscule quantities that they are irrelevant for further investigation.
RNA-Seq based analyses offer considerable power in investigating tumour and stromal responses to anti-angiogenic therapies with human tumour in mouse xenograph models, improving understanding of the effect these therapies have on the tumour environment. Widely used array-based systems profile the transcriptome expression through the use of probes complementary to transcripts from any known gene in the genome. Thus being a semi-targeted approach it suffers from biased transcript coverage17, but also is often unable to differentiate between transcripts derived from homologous genes in different species. Bradford et al. (2013)17 demonstrated how RNA-Seq can be used to dissect transcripts derived from the host (mouse) tissue and the tumour (human) tissue through a species-specific mapping approach, which allowed them to independently investigate tumour and stromal responses to AstraZeneca’s investigational anti-angiogenic drug cediranib (Figure 1).
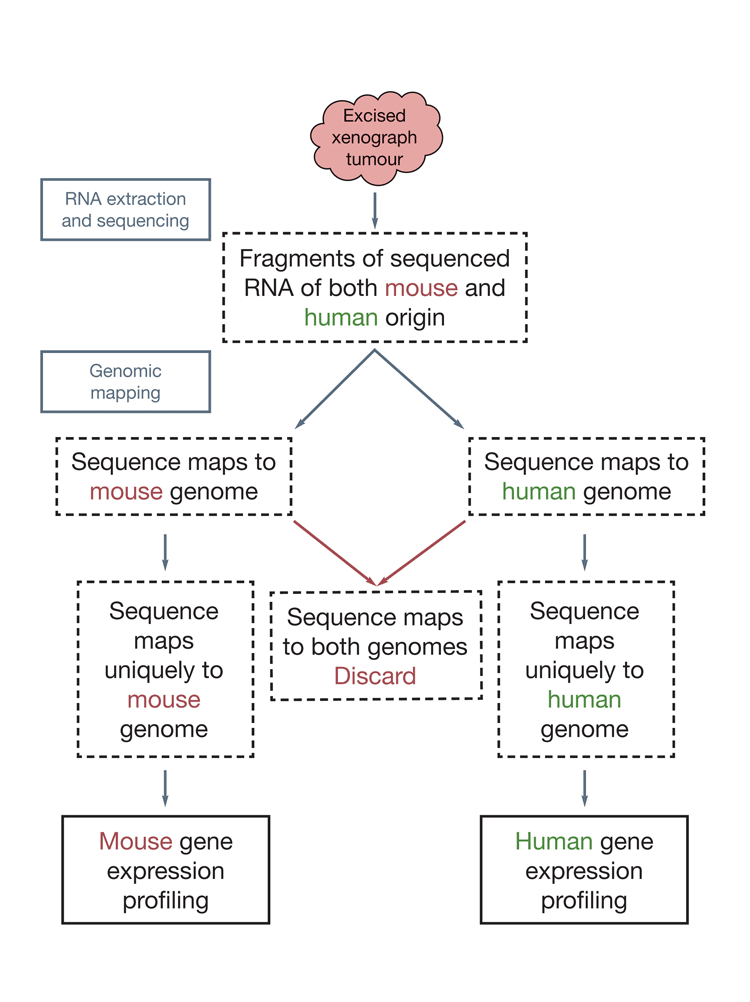
Figure 1: Dissection of human tumour and mouse stromal responses in a xenograph model by RNA-Seq. The human Calu-6 non-small cell lung cancer cell line was grown in mouse. The tumour was excised, the RNA extracted and subjected to RNA-Seq. The sequence read fragments were aligned to a human and mouse reference genome and reads that mapped uniquely to either genome were used for gene expression profiling of both the mouse- and human-derived tumour compartments.
RNA-Seq to study vascular targeted therapies
Vascular disrupting therapies target differences in the vasculature supplying the tumour and healthy tissues. These are either structural differences, such as combretastatin-based therapies, which destabilise the vessels18, or molecular changes that have occurred in the endothelial cells lining the vessels, such as vascular-targeted therapies (VTAs), which aim to specifically kill these cells in a targeted manner12. For VTAs to be effective, targets must be found that are uniquely over expressed on tumour vessels, so that therapeutics can be engineered to target these molecules, localise to the tumour and deliver a toxic payload, killing the vessels. Burrows and Thorpe 199319 were the first to demonstrate the power of this approach. They engineered a neuroblastoma tumour to express interferon gamma and this induced the expression of MHC-class II on the surface of the vessels supplying the tumour. They treated the tumour with the intravenous inoculation of an antibody targeted against MHC class II and conjugated to ricin. This resulted in rapid haemorrhagic necrosis within the tumour, which became completely necrotic over a period of nine days19 (Figure 2).
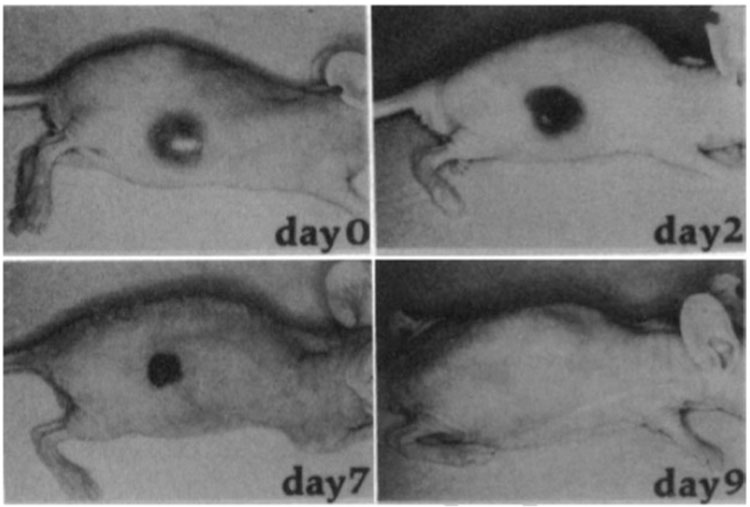
Figure 2: Vascular targeted therapies induce tumour necrosis. Gross appearance of a subcutaneous neuroblastoma tumour treated with a MHC class II targeted immunotoxin. At day 0 the tumours appear highly vascular. Two days after treatment, the tumour is blackened (indicating massive intratumoural haemorrhage). At day 7 the tumour has collapsed into a scab-like plug and by day 10 there is no visible living tumour (figure adapted from 19)
The search for specific tumour vascular targets is an active area of investigation with many groups using transcriptomic analysis to probe for molecular differences between the vessels supplying the tumour and healthy tissues20-23(reviewed in 10). Again, the majority of this work has been performed using current generation technologies, however NGS is starting to be used to provide a more in-depth analysis of the tumour vessel transcriptome.
Zhuang et al. (2013)24 used a combined approach of both microarray and RNA-Seq NGS transcriptomic profiling of isolated endothelial cells from lung cancer. It has been reported that there is only moderate concordance between differentially expressed genes identified by RNA-Seq and by microarray, probably due to the considerable inherent differences between the approaches1. Both approaches suffer from high false positive rates25,26. By using the current and next-generation technologies concurrently, the selection process for candidate genes in lung cancer was streamlined, since the true differences between the healthy and cancer vessels should be identified by both approaches24.
One approach used to identify novel markers of tumour endothelium is to model the environment to which the tumour vessels are exposed. The tumour often grows at a rate exceeding the capacity of the vessel bed to expand and supply it sufficiently27. Because of this, the tumour microenvironment is generally highly hypoxic, hypoglycaemic and acidic with poor blood flow. The vessels are additionally often disordered and poorly structured, leading to breakages within the vessel wall and opening a thrombogenic surface on which clots can form28. This environment has a considerable impact on the expression profile of tumour vessels, creating distinct changes that can be targeted therapeutically.
Zhang et al. (2012)29 modelled the impact exposure to thrombin has on gene expression in pulmonary microvessels, in cancer and other diseases, by RNA-Seq transcriptomic profiling. This analysis identified 150 known genes and 480 known isoforms which were upregulated and 2,190 known genes and 3,574 known isoforms downregulated by exposure to thrombin by at least two-fold. Of note however, is that 1.775 previously unknown isoforms were found to be upregulated and 12.202 downregulated29, highlighting another considerable advantage provided by the next-generation approach. As discussed, microarray-based systems rely on a finite number of predesigned probes to assess the transcript content of a sample, meaning that it is only ever possible to compare the expression of those genes and isoforms for which probes have been generated. On the other hand, by sequencing the entire sample and providing a readout for each transcript encountered, RNA-Seq provides virtually unlimited genomic analysis capacity, allowing the identification of previously unknown genes, isoforms and splice variants1. NGS therefore offers a huge potential for the identification of previously unknown candidates for tumour vascular-specific targeting.
The complexity of RNA-Seq analysis
The generation of such in-depth genomic data is not without its disadvantages. Gigabytes of data are produced for each sample analysed, making the handling of large data sets from multiple samples a considerable and costly challenge. Additionally, bioinformatic tools to assure sequence quality, conduct sequence alignment and assembly and biologically interpret the data are in their infancy, and require considerable development in order to allow NGS to fully achieve its potential for genomic and transcriptomic discovery. The use of these tools to analyse NGS data is costly due to its requirement for considerable investment in infrastructure, but also due to its need for specialist bioinformaticians. The analysis process is also labour- and time-intensive. Firstly, the images from the NGS sequencers must be analysed and converted into sequence reads. The reads are then quality assessed and aligned to a reference genome, if one exists. Finally, mapped and unmapped reads are assessed through a process of gene expression profiling1 (Figure 3). This process of data analysis is considerably more complex and costly than for array-based systems30.
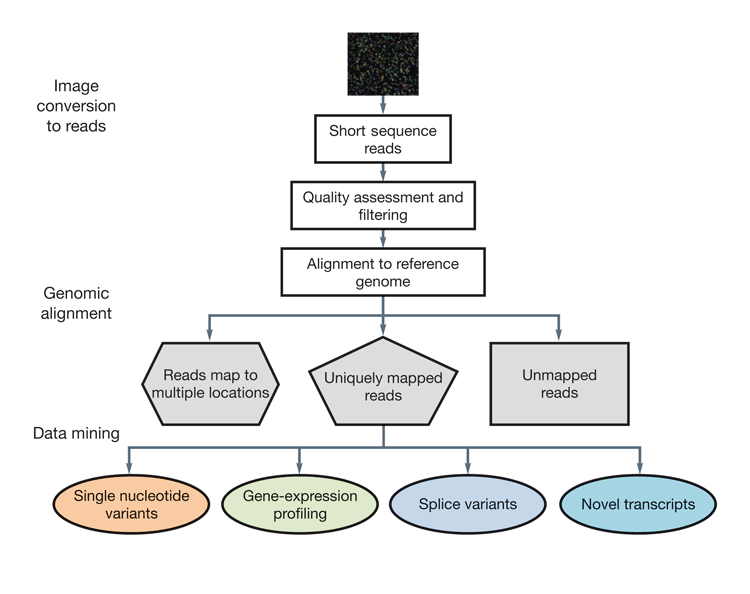
Figure 3: A typical workflow for the analysis of RNA-Seq data. Consists of the conversion of raw data images to short reads, the quality assessment of the reads, alignment to a reference genome (if available) and production of useful data from uniquely mapped reads.
Therefore, despite the rapidly falling costs of performing NGS, the difficulties of analysing the data make it an unattractive option for all but the best-funded research groups, compared with cheap and easily analysed array-based systems. This issue must be addressed with institutional investment in infrastructure, improvement in the quality and usability of analysis software and education in how to use it. The most intelligently designed experiments and the best analysis is achieved when researchers themselves can perform it rather than relying on specialist bioinformaticians unfamiliar with the research area for which NGS is being used. Until the usability of NGS analysis software is improved to the level at which microarray analysis operates, the utility of NGS to enable scientific discovery will be curtailed.
Conclusion
In conclusion, NGS is an extremely promising new approach to enable scientific discovery in the field of tumour vascular biology and in the future could transform our ability to both research and treat cancer. The MD Anderson genetic profiling of abnormal responders is an early example of the potential of NGS to permit personalised medical care. With time, databases for disease-specific mutations and alterations will become more comprehensive and better characterised, allowing NGS of patient material to become a viable option for clinical as well as scientific evaluation, superseding traditional diagnostic methods, which are limited by their narrow focus and usually only useful for one intended purpose.
Acknowledgements
The authors would like to thank Cancer Research UK for financial support and members of the Bicknell/Heath group for intellectual support.
References
- Su Z, Ning B, Fang H, Hong H, Perkins R, Tong W, et al. Next-generation sequencing and its applications in molecular diagnostics. Expert Rev Mol Diagn. 2011 Apr;11(3):333–43
- Harris TD, Buzby PR, Babcock H, Beer E, Bowers J, Braslavsky I, et al. Single-molecule DNA sequencing of a viral genome. Science. American Association for the Advancement of Science; 2008 Apr 4;320(5872):106–9
- Chaisson MJ, Pevzner PA. Short read fragment assembly of bacterial genomes. Genome Res. Cold Spring Harbor Lab; 2007 Dec 1;18(2):000–0
- Yi X, Liang Y, Huerta-Sanchez E, Jin X, Cuo ZXP, Pool JE, et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science. American Association for the Advancement of Science; 2010 Jul 2;329(5987):75–8
- Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nature Biotechnology. Nature Publishing Group; 2008 Dec 1;26(12):1351–9
- Hiller D, Jiang H, Xu W, Wong WH. Identifiability of isoform deconvolution from junction arrays and RNA-Seq. – PubMed – NCBI. Bioinformatics. Oxford University Press; 2009 Nov 17;25(23):3056–9
- Voelkerding KV, Dames SA, Durtschi JD. Next-generation sequencing: from basic research to diagnostics. – PubMed – NCBI. Clinical Chemistry. American Association for Clinical Chemistry; 2009 Mar 26;55(4):641–58
- Folkman J, Cole P, Zimmerman S. Tumor behavior in isolated perfused organs: in vitro growth and metastases of biopsy material in rabbit thyroid and canine intestinal segment. Annals of Surgery. Lippincott, Williams, and Wilkins; 1966 Sep;164(3):491–502
- Folkman J. Tumor Angiogenesis: Therapeutic Implications. N Engl J Med. 1971 Nov 18;285(21):1182–6
- Wragg JW, Bicknell R. Vascular Targeting Approaches to Treat Cancer. Cancer Targeted Drug Delivery. New York, NY: Springer New York; 2013. pp. 59–95
- Folkman J. Anti-angiogenesis: new concept for therapy of solid tumors. Annals of Surgery. Lippincott, Williams, and Wilkins; 1972 Mar;175(3):409–16
- Thorpe PE. Vascular targeting agents as cancer therapeutics. Clin Cancer Res. American Association for Cancer Research; 2004 Jan 15;10(2):415–27
- Hayden EC. Cutting off cancer’s supply lines. Nature. Nature Publishing Group; 2009 Apr 8;458(7239):686–7
- Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nature Publishing Group. 2008 Aug;8(8):592–603
- Subbiah V, Meric-Bernstam F, Mills GB, Shaw KRM, Bailey A, Rao P, et al. Next generation sequencing analysis of platinum refractory advanced germ cell tumor sensitive to Sunitinib (Sutent®) a VEGFR2/PDGFRβ/c-kit/ FLT3/RE… – PubMed – NCBI. J Hematol Oncol. BioMed Central Ltd; 2014;7(1):52
- Meric-Bernstam F, Farhangfar C, Mendelsohn J, Mills GB. Building a personalized medicine infrastructure at a major cancer center. – PubMed – NCBI. Journal of Clinical Oncology. American Society of Clinical Oncology; 2013 May 17;31(15):1849–57
- Bradford JR, Farren M, Powell SJ, Runswick S, Weston SL, Brown H, et al. RNA-Seq Differentiates Tumour and Host mRNA Expression Changes Induced by Treatment of Human Tumour Xenografts with the VEGFR Tyrosine Kinase Inhibitor Cediranib. Müller R, editor. PLoS ONE. 2013 Jun 19;8(6):e66003
- Nihei Y, Suzuki M, Okano A, Tsuji T, Akiyama Y, Tsuruo T, et al. Evaluation of antivascular and antimitotic effects of tubulin binding agents in solid tumor therapy. Jpn J Cancer Res. 1999 Dec;90(12):1387–95
- Burrows FJ, Thorpe PE. Eradication of large solid tumors in mice with an immunotoxin directed against tumor vasculature. Proc Natl Acad Sci USA. 1993 Oct 1;90(19):8996–9000
- Herbert JM, Stekel D, Sanderson S, Heath VL, Bicknell R. A novel method of differential gene expression analysis using multiple cDNA libraries applied to the identification of tumour endothelial genes. BMC Genomics. 2008;9(1):153
- Masiero M, Simões FC, Han HD, Snell C, Peterkin T, Bridges E, et al. A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell. 2013 Aug;24(2):229–41
- St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, et al. Genes expressed in human tumor endothelium. Science. 2000 Aug 18;289(5482):1197–202
- van Beijnum JR. Gene expression of tumor angiogenesis dissected: specific targeting of colon cancer angiogenic vasculature. Blood. 2006 Oct 1;108(7):2339–48
- Zhuang X, Herbert JMJ, Lodhia P, Bradford J, Turner AM, Newby PM, et al. Identification of novel vascular targets in lung cancer. Br J Cancer. 2014 Dec 23;112(3):485–94
- Pawitan Y, Michiels S, Koscielny S, Gusnanto A, Ploner A. False discovery rate, sensitivity and sample size for microarray studies. Bioinformatics. Oxford University Press; 2005 Jul 1;21(13):3017–24
- González E, Joly S. Impact of RNA-seq attributes on false positive rates in differential expression analysis of de novo assembled transcriptomes. BMC Res Notes. BioMed Central Ltd; 2013;6(1):503
- Tannock IF. Population kinetics of carcinoma cells, capillary endothelial cells, and fibroblasts in a transplanted mouse mammary tumor. Cancer Res. American Association for Cancer Research; 1970 Oct;30(10):2470–6
- Falanga A. The Cancer-Thrombosis Connection. The Hematologist. 2011 Jul 1
- Zhang LQ, Cheranova D, Gibson M, Ding S, Heruth DP, Fang D, et al. RNA-seq Reveals Novel Transcriptome of Genes and Their Isoforms in Human Pulmonary Microvascular Endothelial Cells Treated with Thrombin. Zhu D, editor. PLoS ONE. 2012 Feb 16;7(2):e31229
- Hegde P, Qi R, Abernathy K, Gay C, Dharap S, Gaspard R, et al. A concise guide to cDNA microarray analysis. BioTechniques. 2000 Sep;29(3):548–50–552–4–556passim
Biographies
Joseph W. Wragg carried out his undergraduate studies at The University of York and is currently studying for his PhD at the University of Birmingham. He has published three peer reviewed articles in the fields of cancer microenvironment and cancer vascular targeting. He has a particular interest in the use of high-throughput gene expression profiling techniques to identify specific genes for cancer targeting.
Roy Bicknell carried out his undergraduate and doctoral studies at Oxford University and his postdoctoral training at Harvard Medical School. He was formerly Professor of Cancer Cell Biology at Oxford and is currently Professor of Cancer Biology and Genomics at the University of Birmingham. He has published over 200 peer reviewed articles in the fields of endothelium and angiogenesis and a particular interest is the difference between tumours compared to healthy tissue endothelium.
Related topics
Next-Generation Sequencing (NGS)
Related conditions
Cancer
Related organisations
Birmingham University
Related people
Joseph Wragg, Roy Bicknell