The challenges associated with ‘de-risking’ early-stage therapeutic targets
Posted: 3 December 2015 | Ahmad Kamal (MRC Technology), Catherine Kettleborough (MRC Technology), Debbie Taylor (MRC Technology), Tim Chapman (MRC Technology) | No comments yet
The pharmaceutical industry is searching for novel drug targets that could produce the next generation of ‘first-in-class’ therapeutic agents. The challenge for academia is to identify and translate such targets and to provide starting points and confidence in the underlying science. The aim should be to generate evidence that modulation with chemical or biological modulators is likely to result in clinical benefit. As a ‘not-for profit’ drug discovery organisation, MRCT is just one of a number of groups in the UK that conducts translational and drug discovery work in collaboration with academics (http://ukddc.org). Here, we review some of the issues related to working on innovative drug targets, reflect on our own experiences of working in the early-stage drug discovery arena and discuss approaches that allow target ‘de-risking’ and validation for progression into the drug discovery pipeline.

Early-stage innovative drug targets
The identification of protein targets that have genuine therapeutic potential is a major challenge. Although there is wide variation, from our experience over the past 10 years, most new targets that are proposed by academics to translational groups have limited disease association and target validation data to progress them as candidates for drug discovery. These data are not limited to, but may typically include, a combination of evidence including: (i) in vivo or cellular gene silencing or knock-out/knock-in data to indicate a significant change in phenotype; (ii) evidence that gain- or loss-of-function mutations or changes in gene expression are observed in patient populations; (iii) data from the use of non-specific tool compounds that cell signalling is altered or a functional effect or change in phenotype is observed in a cellular or in vivo model; (iv) evidence to show interaction with specific receptors or protein complexes and involvement in cellular processes or signalling events.
Often the data have not yet been published or reproduced by any other research groups. It is also common for the target recombinant protein, essential for the development of screening assays, structural studies and immunisations, to be unavailable, available in only limited amounts or of inadequate quality. In addition, it is unusual for robust target-specific biochemical, biophysical or phenotypic cellular assays amenable to pharmacological or high-throughput screening to be available, further adding to the challenges of progression into a drug discovery program.
The main challenge as we see it and one of the most common reasons for failure of projects is target validation and linked to this is the availability of tool molecules that can be used for conducting such studies, as well as the availability of reagents and assays to enable the identification of such tools. We have learnt that at the beginning of any drug discovery project, it is important that a thorough review is conducted around the potential new target and that key data is both independently reproduced and expanded upon prior to embarking on expensive and time-consuming work to identify tools and/or potential drugs. The target review should encompass a comprehensive review of the scientific, patent and clinical literature, as well as bioinformatic databases to establish a robust target rationale and freedom to operate, as well as determining the availability of reagents, tool compounds/antibodies, biochemical and cellular assays, and in vivo models. It is also vital at this stage that a clear strategy for target validation, identification of modulators, and the generation of reagents and assays should be established, by asking some key questions (see Table 1).
Table 1. Target validation questions that should be considered prior to working on new targets
|
Key Questions |
|
Target Related |
|
Is the target expressed in relevant human cells/tissues? |
|
Is there a disease/diseases associated with the target based on clinical observations? |
|
If the association is genetic, what evidence is there that there is equivalent modification of protein levels in response to disease status and outcome? |
|
Should the target be blocked or activated? |
|
Are there any existing modulators used clinically or available commercially or identified by originating lab? |
|
If target validation is based on knockdown/deletion studies, how well controlled are they? Are they complimented by small molecule or antibody studies that show the same phenotype? |
|
Is there a relevant in vivo model of the disease(s)? How well does it “model” or predict human disease? |
|
Can the target be assayed in a relevant cell-based or in vivo system? How robust is this assay? |
|
Can we access primary human material to investigate the biology of the target (expression, function)? |
|
Is there a phamacodynamic biomarker directly linked to target activity? How easy is it to assay? |
|
Phenotype Related |
|
What is the cellular/physiological process being modulated? |
|
Is it relevant to disease and supported by clinical observations? |
|
Can it be modelled in vitro? In what cellular system? |
|
Can it be modelled in vivo? In what system |
Addressing the challenges
Generation of proteins and reagents for assay development
To date, our focus at MRCT has very much been target-based drug discovery and as such the availability of high quality target protein for the development of assays, as an antigen for immunisation, for structural work, or for target engagement studies, has been critical. Although cellular assays, where the target protein is either recombinantly or endogenously expressed, can be used as a means to screen for modulators, ideally a source of purified target protein is required for biochemical and biophysical assays. Unfortunately, and particularly when working on more novel and unprecedented targets, protein availability, i.e., expression, purification and stability, can be a challenge.
While in some instances commercial sources may be available, this can prove expensive, particularly if the project progresses into a full drug discovery programme. In addition, careful consideration should be given to protein from commercial sources; which expression system has been used? Is it full length or truncated? How has it been purified and what is the level of purity? Is there evidence it is functional? How has this activity been assayed? In prosecuting early stage projects this is a problem that we have encountered on numerous occasions and significant commitment of resource is required to generate active/functional protein in the required amounts.
In some instances, protein stability has been an issue and fresh batches of protein have been required on a regular basis1. In order to address this problem, we invest early to ensure target proteins and antibodies are “fit for purpose” for target validation studies and then onward into a drug discovery programme. As well as establishing in-house protein expression (prokaryotic and eukaryotic), we endeavour to achieve high levels of purity and routinely assess aggregation, solubility and stability.
Target validation
Ultimately, target validation can only be achieved in the clinic, however, robust disease-linkage and modulation of cellular phenotypes in physiologically-relevant assays can contribute to mitigating attrition. To conduct thorough target validation studies, access to bioinformatics and good quality selective tool molecules/antibodies are important. Equally vital is the ability to confirm engagement of the modulator with the target, especially in a cellular format. Unfortunately, such tools or information may not be readily available, requiring a combination of alternative approaches to achieve confidence in the target. In our experience it is vital to start by asking key target-related and phenotype-related questions (see Table 1) and put together a plan of the required studies. We should not be afraid to “fearlessly evaluate the science”2.
Target validation can involve a combination of many different techniques including amongst others: various methods for gene editing/silencing, the use of various forms of biological and chemical tools and the use of multiple types of animal models (see Figure 1). Where possible target validation studies can be supported by the use of patient-derived or primary human cells/tissues or physiologically relevant cell lines, such that species differences and artificial systems do not impact on the results obtained and effects can be translated into the clinic. The use of phenotypic and high-content assays, and the use of cells generated from human induced pluripotent stem cells also have a role.
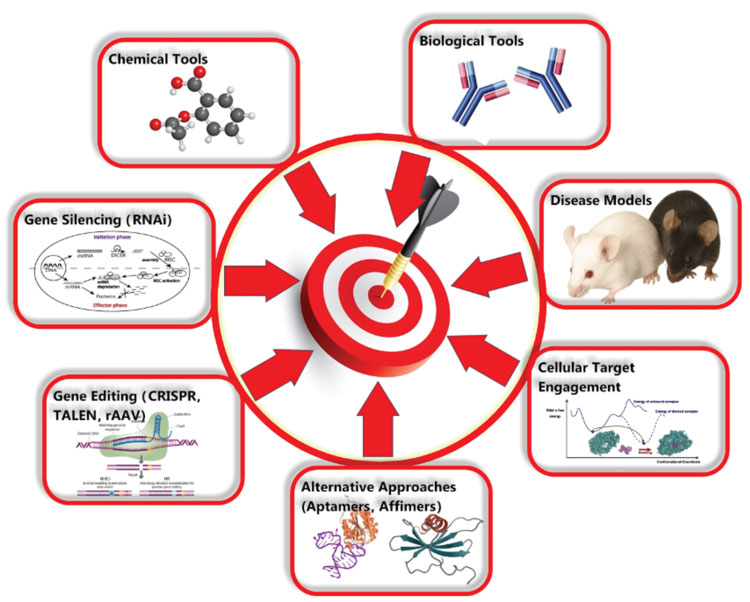
Figure 1: Techniques that can be used for target validation
It is essential to have confidence in the authenticity of any tools, reagents, antibodies and cell lines used for target validation work and ensure that they are ‘fit for purpose’. In-house generation of protein and characterisation will address some of these issues, however, other things to consider include: authentication and mycoplasma-testing of any cell lines used; whether antibodies from commercial or external sources are being used as part of target validation studies; how they were generated and, depending on the studies they will be used for, are there preservatives?
Another question to ask is: are they endotoxin-free? Since target validation studies can involve complex experiments, in our experience investing in the characterisation of reagents and tools will save time and resources, as well as give confidence in the results obtained from such studies. We consider it important that any published or reported data is robustly reproducible for “landmark experiments”, but unfortunately in our experience, and as others have reported, not all published data stands up to scrutiny2,3,4.
At MRCT we have prosecuted more than 83 projects over the past 10 years and an analysis of the data indicates that 25% of these programs were closed early due to lack of target validation. In a significant number of these programs, the data we generated, particularly where we could access pharmacological modulators, did in fact help to ‘de-validate’ targets. Based on this experience, we are now putting greater emphasis and resource into early target validation work and are establishing powerful new technologies ‘in house’ that can be used for such studies including: CRISPR gene silencing/editing and cellular thermal shift assays. Overall, we aim to fulfil three criteria during the project lifecycle, encompassing: (i) evidence that the target is associated with disease; (ii) that the modulator interacts with the target; and (iii) that the modulator results in a relevant phenotype, ideally in relevant human cells.
MRCT also recognises the advantage of working in collaboration with other groups, particularly in relation to sharing of target validation data. We have numerous collaborations with pharmaceutical companies worldwide on specific targets and play a key role in therapeutically focussed initiatives with charitable organisations (e.g., Alzheimer’s Research UK Dementia Consortium)5.
Generation of Tools
It has become increasing clear that the availability of tools, including both antibodies and selective small molecules, has a significant impact on the ability to validate a drug target and at MRCT we recognise the importance of generating such tools during the early stages of a programme. We do this via a combination of ‘in-house’ synthesis, commercially-available compounds or expression of antibodies described in the scientific or patent literature, as well as via screening programs to identify novel modulators.
Antibodies
In addition to the traditional approach of generating monoclonal antibodies via the immunisation of rodents and screening of hybridomas, we are increasingly using phage library generation and screening from immunised sources (mouse, rabbit llama) to generate scFvs or nanobodies (single heavy chain variable regions) as tools for target validation. If required these formats can readily be converted to whole IgGs or other isotypes if applicable. The ideal criteria for an antibody tool are shown in Table 2, but key is that it demonstrates selective nanmolar binding affinity and can be used to obtain functional effects when used at ~1µg/ml (~7nM). For target validation studies it needs to be soluble, endotoxin–free, and of a relevant isotype. Nanobodies are generally easy to express and purify and give high yields from either mammalian cells or E.coli. They have proved to be useful tools for target validation, for affinity purification of proteins, as well as for protein stabilisation to aid structural studies.
Table 2. Ideal criteria for an antibody tool
|
Criteria |
|
|
Affinity |
Min 1-10 nM |
|
Functional activity (this is the key criteria) |
Observed at concentrations <1ug/ml |
|
Solubility |
Better than 10 mg/mL in PBS |
|
Biophysical properties |
No aggregation, not sticky, purifies under standard conditions |
|
Stability |
Stable at 4oC for more than 1 month |
|
Cross-reactivity |
Project dependent: Needs to bind to species required for assays. For a therapeutic: Human and cynomologus binding with preferred rodent binding. Alternatively, an orthologue rodent antibody also developed. |
|
Known epitope/ sequence |
Not necessary for a tool |
|
Effector function |
Formatted as appropriate |
Small molecules
The ideal criteria for small molecule tools or probes are shown in Table 3. With the unprecedented nature of the targets that we prosecute at MRCT, typically there are no existing probes available. Where compounds are available, even if they meet potency criteria, they are often not sufficiently characterised or do not meet the desired quality criteria in other areas, such as selectivity or physicochemical properties and sometimes they contain known frequent hitter motifs. High-quality probes are essential for performing target validation work and therefore these have to be generated. We find that it is important to have a range of compound sets available for screening as potential start points, so the hit generation approach can be tailored depending on the project. These sets include annotated known bioactives, target-class based sets and diversity-based sets spanning fragment, lead-like and drug-like chemical spaces, as well as pathway-focused sets to support phenotypic approaches. Active curation of these is important and we have recently expanded our fragment set, renovated our diversity set and are currently renewing our kinase-focused set.
Table 3. Ideal criteria for small molecule tools/probes
|
Criteria |
|
|
Structure |
Well characterised; no reactive groups unless a selective mechanistic requirement |
|
Stability |
High chemical stability in aqueous media |
|
Biochemical activity |
<100nM |
|
Cellular activity |
<1µM; concentration-dependent effect observed |
|
SAR |
Closely related structures with similar activity |
|
Selectivity |
>100-fold versus anti-target/s; polypharmacology undesirable |
|
Permeability |
High |
|
Solubility |
High (>100 µM) in low % DMSO aqueous buffered solutions, no aggregation effects |
|
Negative control compound |
Inactive compound with similar structure shows no activity in cells |
|
Orthogonal probes |
Different chemotypes with similar levels of activity/selectivity |
|
Mechanism of action |
Well-defined quantitative relationship between biochemical and cellular effects consistent with target-dependent action |
|
Pharmacokinetics |
Good PK not essential for in vitro and cellular work, but required for animal work |
|
Toxicity |
No undesired effects |
As noted elsewhere6,7,8, the requirements for a chemical probe are somewhat different from those of a drug. Some parameters are less rigorous for a probe, for example, tools for cellular studies will not require optimisation of pharmacokinetic (PK) parameters. However, other requirements such as selectivity are more stringent for a probe than a drug, since the polypharmacology that may be advantageous for the efficacy of a drug is undesirable in a probe, because it does not allow the target to be confidently linked with the phenotype. It is recommended that different chemotypes with similar levels of activity are used for validation work, as well as control compounds that are inactive, although in practice for novel targets at MRCT it can be difficult to generate multiple active series with the right profiles. Importantly, the impact of a probe in elucidating the biology will partly depend on its ready and widespread availability6, and at MRCT we endeavour to make tools available to academics.
An evaluation of projects conducted over the past 10 years indicates that tool compounds were generated and made available for 38% of small molecule and 57% of antibody projects. Notably, a new resource to share information around probes has just been launched (http://www.chemicalprobes.org/). We are also making increased use of bespoke tagged probes alongside the development of SAR in a chemical series, to increase confidence in the relationship between engagement of the target and the observed phenotype. These include biotinylated probes to allow identification or confirmation of the compound interaction partners in the cell; we typically conduct the proteomics analysis work in collaboration with academic partners9,10 and intend to build our capabilities in this area to examine target engagement in intact cells as well as lysates.
Conclusion
Our experiences of working as a translational unit at the interface between academia and pharma over the past 10 years has taught us that early investment in validation of targets is essential for successful management of a portfolio of novel and unprecedented targets. Historically, a significant number of our projects have failed due to lack of target validation and on some occasions projects have stalled because of issues with protein generation or an inability to identify validated hits. To address this we are putting a greater emphasis on generation of reagents and tools fit for purpose, increased the use of new technologies and are repeating key studies to confirm the proposed therapeutic hypothesis. Coupled with a rigorous target selection procedure we envisage a greater success rate going forward.
References
- McNeil, EM, Ritchie, AM, Astell, KR, Shave, S; Houston, DR, Bakrania, P., Jones, HM, Khurana, P, Wallace, C, Chapman, T, Wear, MA, Walkinshaw, MD, Saxty, B and Melton, DW. Inhibition of the ERCC1-XPF structure-specific endonuclease to overcome cancer chemoresistance. 2015; DNA Repair 31: 19-28
- Dahlin, JL, Inglese, J and Walters, MA. Mitigating risk in academic preclinical drug discovery. Nat. Rev. Drug Discov. 2015; 14:279-294
- Prinz, F, Schlange, T and Asadullah, K. Believe it or not: how much can we rely on published data on potential drug targets? Nat. Rev Drug Discov. 2011; 10: Correspondence
- Mullard A. Reliability of ‘new drug target’ claims called into question. Nature Rev Drug Discov. 2011; 10: 643
- Bryans, J. ( 2015). A new dawn in academic and pharma collaboration – a UK perspective. Drug Target Review, 2: 12-14
- Frye, SV. The art of the chemical probe. Nat. Chem. Biol. 2010; 6: 159-161
- Workman, P and Collins, I. Probing the probes: Fitness Factors for Small Molecule Tools. Chemistry & Biology. 2010; 17: 561-577
- Bunnage, ME, Piatnitski Checkler, EL and Jones, LH. Target validation using chemical probes. Nature Chem Biol. 2013; 9: 195-199
- Tsaytler, P, Harding, HP, Ron, D and Bertolotti, A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science. 2011; 332: 91-94.
- Green, J, Moon, R, Whalley, D, Bowyer, P, Wallace, C, Rochani, A, Kumar, R, Howell, S, Jones, H, Ansell, K, Chapman, T, Taylor, D, Osborne, S, Baker, D, Tatu, U and Holder, A. Imidazopyridazine inhibitors of Plasmodium falciparum calcium dependent protein kinase 1 also target cGMP-dependent protein kinase and heat shock protein 90 to kill the parasite at different stages of intracellular development. Antimicrob. Ag. Chemother. 2015. Submitted
Biographies
Ahmad Kamal is a Team Leader in MRCT’s Cellular Pharmacology group, establishing cell-based assays for target validation and profiling of both small molecules and antibodies. He has worked in the pharmaceutical industry as well as academia, having graduated with a PhD in Pharmacology from Imperial College.
Debra Taylor is a Drug Discovery Manager at MRCT’s Centre for Therapeutics Discovery. She is responsible for managing the team responsible for target validation and conducting cell-based assays to support small molecule and antibody programs. She obtained her PhD from University of London and previously worked in antiviral drug discovery.
Tim Chapman is a Team Leader in chemistry at MRC Technology’s Centre for Therapeutics Discovery, where he has worked on discovery projects across a range of disease areas including malaria, TB, oncology and inflammation. He obtained a DPhil in chemistry at Oxford University, followed by a postdoctoral fellowship at the University of Toronto.
Catherine Kettleborough is the Associate Director of Biology at MRC Technology’s Centre for Therapeutics Discovery (CTD). CTD’s Biology group is responsible for conducting target validation, assay development, screening and cell based assays to support prosecution of early drug discovery projects for novel therapeutic targets sourced from academic research groups worldwide.
Related topics
Assays, Drug Discovery, Drug Targets, Therapeutics
Related organisations
MRC Technology
Related people
Ahmad Kamal, Catherine Kettleborough, Debbie Taylor, Tim Chapman